- Teste de hematologie
- Teste de biochimie
- Biochimie generală din sânge și urina
- Proteine specifice in ser si urina
- Teste biochimice din lichide de punctie
- Teste biochimice din materii fecale
- Teste biochimice pentru tulburari ereditare de metabolism
- Teste pentru nefrolitiaza
- Vitamine, oligoelemente, stres oxidativ
- Acizi grași
- Transferina carbohidrat deficitara (CDT) marker pentru alcoolism
- Markeri non-invazivi pentru afecţiunile hepatice
- Analiza chimică calculi
- Markeri endocrini
- Markeri tumorali
- Markeri virali
- Markeri cardiaci
- Markeri anemie
- Markeri ososi
- Markeri boli autoimune
- Anticorpi antispermatozoizi
- Autoanticorpi in afectiuni endocrine, cardiace, renale
- Autoanticorpi in afectiuni neurologice
- Autoanticorpi in afectiunile dermatologice
- Autoanticorpi in anemia pernicioasa
- Autoanticorpi in diabetul zaharat
- Markeri pentru afectiuni hepatice si gastrointestinale autoimune
- Markeri pentru afectiuni reumatismale si vasculite
- Markeri pentru monitorizarea evolutiei si tratamentului
- Markeri pentru sindromul antifosfolipidic
- Serologie boli infectioase
- Teste specializate de alergologie si imunologie
- Teste de biologie moleculara
- Teste de citogenetica
- Teste de microbiologie
- Toxicologie
- Citologie cervico-vaginala
- Histopatologie
- Consult genetic
- Genetica medicala

JAK2 – mutații exon 12&14 (V617F)
Preț: 866.00 lei
Informatii generale – JAK2 – mutații exon 12&14 (V617F)
t Noua nomenclatura a entitatilor mieloproliferative s-a modificat de la “boli mieloproliferative cronice” (MPD, Chronic Myeloproliferative Diseases) la “neoplazii mieloproliferative” (MPN, Myeloproliferative Neoplasms) pentru a reflecta cu acuratete natura lor neoplazica. De asemenea subgrupul desemnat anterior ca “boli mielodisplazice/mieloproliferative” a fost redenumit “neoplazii mielodisplazice/mieloproliferative” (MDS/MPN, Myelodysplastic/Myeloproliferative Neoplasms)11.
Atat MPN cat si MDS/MPN reprezinta neoplazii clonale cu celularitate medulara tipic crescuta, maturatie a liniilor celulare si organomegalie, separarea dintre ele bazandu-se pe prezenta mielodisplaziei in cele din urma. In plus acestea au in comun grade diferite de fibroza medulara, dar difera in general prin linia celulara mieloida care domina hematopoieza.
Dintre MPN exista patru tipuri comune, respectiv leucemia mieloida cronica (CML, Chronic Myelogenous Leukemia), caracterizata prin translocatia 9;22 si proteina de fuziune BCR/ABL1, si trei tipuri non-CML, policitemia vera (PV, Polycythemia Vera), trombocitemia esentiala (TE, Essential Thrombocythemia) si mielofibroza primara (PMF, Primary Myelofibrosis), acestea avand in comun o incidenta crescuta a unei mutatii punctiforme dobandite (V617F) la nivelul JAK2 kinazei, o tirozin kinaza citoplasmatica importanta in proliferarea hematopoietica2. De asemenea au fost identificate si alte mutatii la nivelul caii JAK-STAT la unii pacienti cu MPN JAK2V617F(-), sugerand ca activarea constitutionala a acestei cai de semnalizare este o trasatura comuna acestor afectiuni5.
Practic toate caile de transmitere a semnalului intracelular sunt legate printr-o cascada de fosfotransfer mediat prin kinaze. La om sunt exprimate mai mult de 500 kinaze care fosforileaza proteine distincte, in mod tipic la nivelul reziduurilor de tirozina, serina sau treonina. JAK2 (Janus-associated kinase 2) este membra a unei familii de patru tirozin kinaze citoplasmatice, care include de asemenea JAK1, JAK3 si TYK212.
Tirozin kinazele JAK sunt componente cruciale care integreaza componente din diverse cai de semnalizare intracelulara, incluzand caile Src kinazei, Ras-MAP kinazei, PI3K-AKT si STAT, consecutiv interactiunii receptorilor pentru citokine/interferon cu liganzii lor. Astfel activitatea optima a JAK kinazelor este critica pentru transmiterea normala a semnalului citokinelor si factorilor de crestere.
Supraactivitatea JAK kinazelor a fost implicata in tumorigeneza fiind asociata cu diferite sindroame leucemice, mutatii activatoare in JAK1 fiind recent descoperite in leucemia acuta limfoblastica cu precursor de celula T, in schimb deficienta JAK3 a fost asociata cu imunodeficienta combinata severa si un inhibitor selectiv al JAK3 a fost dezvoltat formand o noua clasa de medicamente imunosupresoare. Deficienta JAK2 la soareci este letala in stadiul embrionar prin esecul eritropoiezei7,12,13.
Structura tridimensionala a JAK kinazelor este in prezent necunoscuta, aceasta datorandu-se si faptului ca acestea sunt proteine relativ mari, de peste 1100 aminoacizi, cu mase moleculare aparente de 120-140kDa. Fiecare din kinazele JAK are 7 domenii bine conservate intre specii, “JAK homology” JH1-JH7, care nu prezinta asemanare cu niciun model proteic cunoscut (vezi figura 1).
Domeniul JH1 de la capatul C-terminal contine domeniul catalitic. Valina din pozitia 617 se afla in domeniul JH2, un domeniu pseudokinazic, care prezinta omologie semnificativa cu domenuiul JH1, dar ii lipseste activitatea catalitica, lipsindu-i cativa aminoacizi critici, necesari pentru o kinaza functionala. Aceasta arhitectura in tandem constituie marca JAK kinazelor si le confera numele, dupa zeul Roman Janus (Ianus) cu doua fete, insemnand inceputul si sfarsitul.
Pe baza observatiei ca deletia domeniului JH2 duce la cresterea activitatii JAK2 kinazei, s-a sugerat ca domeniul JH2 regleaza negativ activitatea kinazica JH15;7;10;13. Capatul N-terminal contine domeniile SH2-like (JH3-JH4) si un domeniu omolog FERM (4-point-1, Erzin, Radixin, Moesin) (JH6-JH7), implicat in interactiunea cu proteine transmembranare, cum ar fi receptorii pentru citokine, si, in plus, leaga domeniul kinazic, reglandu-i pozitiv activitatea catalitica13 (vezi fig. 1).
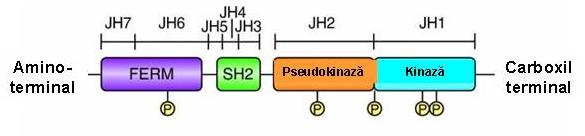
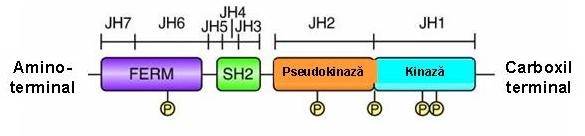
Fig. 1 Reprezentare schematica a structurii primare a Janus-kinazelor (Jaks)
(Adaptare dupa Genome Biol. 2004; 5(12): 253).
Tirozin kinazele JAK sunt constitutiv asociate in stare inactiva cu regiunea juxtamembranara a receptorilor pentru citokine, in unele cazuri interactiunea dintre JAK si receptor fiind crescuta dupa legarea ligandului. S-a propus ca legarea ligandului promoveaza o modificare conformationala a receptorului, care promoveaza activarea JAK prin interactiunea reciproca a doua JAK kinaze juxtapozitionate si auto- si/sau trans-fosforilarea reziduurilor de tirozina in loop-ul de activare al domeniului kinazic13.
In schimb JAK fosforilate mediaza fosforilarea reziduurilor de tirozina ale domeniului citoplasmatic al receptorului si creeaza un situs de docare pentru recrutarea catorva proteine, in final ducand la activarea STAT (signal transducer and activator of transcription), MAP (mitogen-activated protein) kinazei si caii PI3K-AKT. STAT activate dimerizeaza si sunt translocate la nivelul nucleului unde regleaza transcriptia dupa legarea la secvente consens specifice in regiunile promotor ale catorva gene tinta.
JAK2 este de asemenea implicata in expresia receptorilor EpoR (erythropoietin receptor) si MPL (receptorul pentru trombopoietina) pe suprafata celulara actionand ca stabilizator al proteinei. Aceste semnale complexe sunt autonom activate in absenta legarii citokinei la receptorul sau in cazul mutatiilor JAK2 sau ale receptorului (cum ar fi mutatia W515L/K la nivelul MPL)10.
Majoritatea citokinelor se pot asocia cu mai mult de o JAK kinaza, dar JAK2 este esential pentru citokine hormon-like, cum ar fi hormonul de crestere, prolactina, eritropoietina, trombopoietina si familia citokinelor care semnalizeaza prin intermediul receptorului pentru IL-3 (IL-3, IL-5, si GM-CSF), fiind de asemenea important pentru citokine care utilizeaza receptorul gp130 si pentru unii interferoni. JAK2 este JAK kinaza predominanta implicata in proliferarea si diferentierea celulelor mieloide5;13.
Gena JAK2 este mapata la nivelul cromozomului 9p24, fiind codificata de doi produsi de transcriptie, de 5.3 si 5 kb, iar proteina este exprimata ubicuitar, ca si JAK1 si TYK2, spre deosebire de JAK3 care este predominant exprimata la nivelul celulelor hematopoietice7;13.
Mutatia JAK2V617F este o substitutie a guaninei cu timidina la pozitia 1849 in exonul 14 care rezulta in substitutia valinei cu fenilalanina la nivelul codonului 617, care se presupune ca duce la pierderea controlului autoinhibitor al domeniului pseudo-kinazic JH2. Introducerea acestei mutatii la nivelul liniilor celulare duce la cresterea acestora citokin-independenta si la hipersensibilitate la citokine, mimand cresterea in vitro a progenitorilor hematopoietici de la pacientii cu MPN si oferind o explicatie pentru mecanismul formarii coloniilor eritroide endogene in absenta eritropoietinei, care constituie marca PV.
Mutatia nu este prezenta in linia germinala, fiind achizitionata ca o mutatie somatica in compartimentul hematopoietic, descoperita la mai mult de 95% din pacientii cu PV, pana la 50% din cei cu TE si PMF, 40-50% din cei cu anemie refractara cu sideroblasti inelari si trombocitoza (RARS-T), rari pacienti cu CML atipica BCR-ABL1(-), CMML (Chronic Myelomonocytic Leukemia), AML (Acute Myeloid Leukemia) si chiar la unii pacienti cu trombocitoza ereditara sau pacienti normali hematologic cu tromboza de vena porta1;3;5;9;10;11.
Nicio alta mutatie alternativa nu a fost descrisa pana in prezent la acest nivel, alte mutatii activatoare ale JAK2, incluzand T875N la nivelul domeniului kinazic, deletia IREED sau mutatiile exonului 12, fiind mult mai rare comparativ cu JAK2V617F. Pe de alta parte, desi calea de semnalizare JAK2 este constitutiv activata de o varietate de mecanisme genetice si epigenetice in neoplaziile umane, incluzand limfomul si mielomul, alela JAK2V617F este exclusiva pentru neoplaziile mieloide, sugerand mecanisme distincte care activeaza JAK25.
Exista mai multe ipoteze privind contributia unei singure alele de boala la dezvoltarea a trei afectiuni mieloide inrudite, dar distincte clinico-patologic3.
- In prima ipoteza fenotipul depinde de celula tinta a mutatiei JAK2V617F. Studii diferite au aratat ca liniile granulocitara, eritroblastica, megakariocitara si limfoida contin celule JAK2V617F(+) atat in PV cat si in PMF, ceea ce nu exclude insa faptul ca JAK2V617F tinteste subseturi de celule stem hematopoietice diferite, cu programe transcriptionale si proprietati de diferentiere diferite. Intr-adevar exista grupuri care au demonstrat diferente in compartimentul celulelor stem intre PV si PMF3.
- In cea de-a doua ipoteza JAK2V617F este singurul eveniment responsabil de MPN si fenotipul depinde de fondul genetic al pacientului. Din analiza comparativa a datelor din patru studii in care tipuri genetice diferite de soareci au fost transplantati cu celule stem hematopoietice transduse cu JAK2V617F, s-a observat ca acestia reproduc fenotipuri TE, PV sau post-PV, sugerand ca mutatia JAK2 este suficienta pentru a induce MPN3. Al doilea argument privind modularea fenotipului de factori genetici mosteniti vine de la Pardanani si colegii care au gasit ca exista SNP-uri (single nucleotide polymorphisms) specifice in JAK2 si EpoR asociate cu PV sau TE3;5.
- In cea de-a treia ipoteza fenotipul depinde de nivelul activitatii kinazice a JAK2V617F. Mutatia JAK2V617F poate fi gasita fie in stare heterozigota, fie in stare homozigota la pacientii cu MPN, mecanismul care duce la homozigotism fiind in majoritatea cazurilor recombinarea mitotica a cromozomului 93. Astfel colonii eritroide JAK2V617F mutante homozigote sunt observate la majoritatea pacientilor cu PV, chiar la cei cu o incarcatura mica a alelei, dar sunt numai rar observate in TE. Datele cercetarilor sugereaza ca exista diferente calitative si/sau cantitative in semnalizarea constitutiva in celulele JAK2V617F mutante dependente de doza genei, ceea ce afecteaza fenotipul, sugerandu-se ca nivelul semnalizarii JAK2-STAT5 functioneaza ca un reostat care determina fenotipul predominant eritroid sau megakariocitic. Cand este prezenta in stare heterozigota mutatia stimuleaza preferential megakariopoieza, iar in stare homozigota scade megakariopoieza in favoarea eritropoiezei crescute9. In functie de nivelul si durata expunerii activitatea kinazica sustinuta va duce in final la mielofibroza3.
In general cea mai mare incarcatura a alelei V617F, respectiv nivelul alelei mutante fata de alela normala, este gasita la pacientii cu PV, urmati de cei cu PMF si TE1;5,10.
Explicatia pentru modul in care variatii in cantitatea unei proteine mutante unice pot duce la fenotipuri diferite poate proveni din expresia diferita a EpoR, MPL si G-CSFR la nivelul progenitorilor. MPL este exprimat la niveluri crescute in precursorii megakariocitari, sugerand ca o cantitate mica de JAK2V617F ar fi suficienta pentru a induce semnalizarea MPL, proliferarea megakariocitara si productia plachetara, asa cum se observa in TE.
In schimb EpoR este exprimat la niveluri scazute in precursorii eritroizi, astfel cantitati mai mari de JAK2V617F ar fi necesare pentru a induce semnalizarea EpoR si hiperplazia eritroida, ducand la fenotipul de PV. S-a aratat ca suprasemnalizarea MPL prin stimularea excesiva de catre Tpo (trombopoietina) duce la mielofibroza, sugerand ca o cantitate crescuta a JAK2V617F ce duce la o puternica semnalizare MPL in megakariocite ar fi responsabila pentru mielofibroza3.
TE JAK2V617F(+) a fost propusa ca fiind o “forma frusta” de PV pe baza observatiei ca acesti pacienti prezinta nivele mai scazute ale trombocitelor, nivele mai crescute ale hemoglobinei, leucocitelor, nivele mai mici ale eritropoietinei si rate mai mari ale trombozei venoase comparativ cu cei cu TE cu JAK2 normala1;3;9.
In plus pacientii cu PV cu o incarcatura mare de JAK2V617F (>50%) prezinta niveluri semnificativ crescute ale hemoglobinei si o rata crescuta de transformare fibrotica comparativ cu pacientii heterozigoti, iar progresia la mielofibroza post-PV este asociata cu cresterea incarcaturii in JAK2V617F3. Dat fiind faptul ca transformarea mielofibrotica a PV si TE nu poate fi diferentiata de PMF, pacientii diagnosticati cu PMF ar putea reprezenta indivizi care se prezinta intr-o faza accelerata a unei MPN preexistente1. Astfel unii considera MPN JAK2V617F(+) ca fiind o singura boala cu diferite stadii/fenotipuri3;10.
Pe de alta parte, pacientii cu TE JAK2V617F(+) aproape niciodata nu evolueaza la homozigotism, in timp ce pacientii cu PV JAK2V617F(+) aproape intotdeauna au o subclona homozigota care poate fi detectata doar daca se analizeaza colonii individuale. Astfel se considera ca exista o diferenta moleculara fundamentala intre pacientii cu PV si TE mai degraba decat un “continuum” asa cum a fost sugerat de modelul “formei fruste”9.
De asemenea mutatii somatice aditionale pot contribui la patogenia PV, TE si PMF JAK2V617F(+)5.
- In cea de-a patra ipoteza mutatia JAK2V617F poate sa nu fie evenimentul clonogenic initial in dezvoltarea MPN si poate exista o celula mutanta pre-JAK2 care determina fenotipul, in sprijinul acestei afirmatii venind si faptul ca blastii leucemici de la pacientii cu MPN JAK2V617F(+) care evolueaza spre leucemie acuta sunt adesea negativi pentru aceasta mutatie3;10 sau faptul ca pacienti cu MPN JAK2V617F(+) produc colonii eritroide eritropoietin-independente negative pentru mutatia JAK21. Multe grupuri au descris pacienti care prezinta o alta anomalie moleculara asociata JAK2V617F, cum ar fi BCR-ABL, mutatii MPL sau alta mutatie JAK23. Recent au fost raportati pacienti in care achizitia mutatiei JAK2 a fost precedata de deletia cromozomului 20q sau o mutatie in TET2. In plus la o proportie din pacientii cu MPN in faza cronica a fost identificata o boala biclonala in care doua evenimente genetice (mutatii in JAK2 sau MPL sau deletii ale cromozomului 20q) pot fi detectate in expansiuni clonale separate1.
In ultimul rand existenta cazurilor familiale de MPN, in majoritatea lor cu transmitere autosomal dominanta cu penetranta incompleta, este argumentul cel mai puternic pentru prezenta unui eveniment pre-JAK2 la unii pacienti. In toate cazurile JAK2V617F este dobandita ca o alela somatica. In cadrul aceleeasi familii pot coexista entitati clinice diferite si forme JAK2V617F(+) si JAK2V617F(-), sugerand prezenta unui eveniment necunoscut la nivelul liniei germinale ce ar fi un factor predispozant pentru MPN3;5.
Exista studii care demonstreaza ca achizitia mutatiei JAK2V617F este puternic asociata cu mostenirea unui haplotip care include gena JAK2. La heterozigotii pentru acest haplotip mutatia JAK2 a fost observata mult mai frecvent in cis fata de alela predispozanta. Au fost propuse doua modele: modelul hipermutabilitatii, in care alela predispozanta este in mod particular predispusa la achizitia de mutatii ca urmare a unor mecanisme necunoscute, si modelul „terenului fertil”, in care mutatiile JAK2 pot aparea in mod egal la nivelul oricarei alele, dar este mult mai posibil sa rezulte in expansiune clonala cu boala manifesta daca sunt achizitionate in cis cu haplotipul predispozant, poate ca urmare a unei diferente in expresia sau functia proteinei JAK2 codificate de aceasta alela1 (vezi fig. 2).
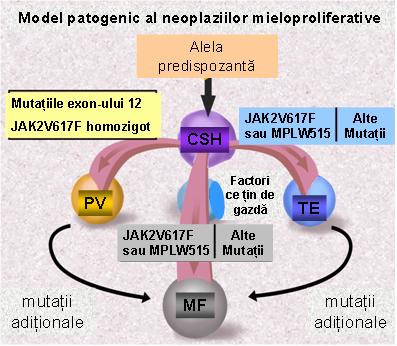
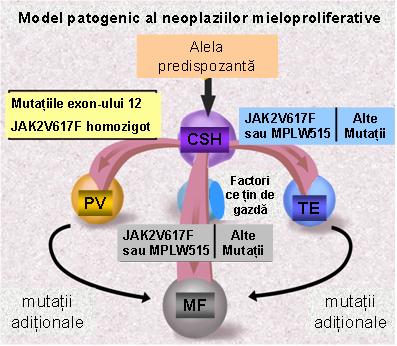
Figura 2 Model pentru patogeneza MPN (Adaptare dupa Levine, R. L. et al. Blood 2008;112:2190-2198).
CHS = celula stem hematopoietica; PV = policitemia vera; TE = trombocitemia esentiala; MF = mielofibroza
Rolul mutatiei JAK2V617F(+) in complicatiile MPN, incluzand tromboza, mielofibroza si transformarea leucemica ramane necunoscut5. Pacientii cu TE JAK2V617F(+) au fost raportati ca prezentand rate crescute de tromboza generala si tromboza venoasa comparativ cu cei fara mutatie, dar o asociere directa intre TE JAK2V617F(+) si evenimentele arteriale nu a fost bine stabilita1.
Studii recente sugereaza ca leucocitoza este un important factor de risc pentru tromboza in MPN, intr-un studiu fiind observata o expresie crescuta a genelor indicative a activarii neutrofilice la pacientii cu ET JAK2V617F(+), sugerand ca exista o relatie intre activarea neutrofilelor, tromboza si semnalizarea JAK2 in MPN5. In ceea ce priveste progresia spre leucemia acuta, aceasta implica achizitia unor alte evenimente genetice.
Studiile efectuate pe linii celulare sugereaza ca expresia JAK2V617F este asociata cu cresterea deteriorarii ADN-ului si reparare aberanta a acestuia. In plus celulele JAK2V617F(+) de la pacienti cu PV sau PMF esueaza in a suferi apoptoza ca raspuns la deteriorarea ADN-ului, furnizand astfel un mecanism pentru acumularea de leziuni genetice. Totusi in jumatate din cazuri leucemia dezvoltata in contextul unei MPN JAK2V617F(+) precedente este negativa pentru mutatia JAK2, existand doua modele alternative pentru a explica acest fenomen: 1. cele doua faze ale bolii sunt legate filogenetic, aparand dintr-o clona fondatoare comuna (pre-JAK2), si 2. MPN si AML sunt nelegate clonal si reflecta transformarea unor celule stem independente1.
Identificarea de mutatii activatoare in JAK2 a dus la dezvoltarea de terapii moleculare tintite pentru acesti pacienti. Trialuri de faza I cu inhibitori JAK2 au fost initiate in PMF si mielofibroza post-PV/TE.
La pacientii cu PV JAK2V617F(+) utilizarea interferonului a fost raportata ca reduce incarcatura alelei mutante, cu obtinerea ocazionala a remisiunii moleculare1.
Spre deosebire de JAK2V617F in care o singura alela a fost identificata, au fost raportate cel putin 8 mutatii distincte in exonul 12 al JAK2, incluzand mutatii missense, deletii, si insertii implicand reziduurile 537-543 (N542-E543del, F537-K539delinsL, E543-D544del, K539L6). Ca si in cazul JAK2V617F, expresia mutatiilor exonului 12 duce la cresterea citokin-independenta a celulelor hematopoietice, activarea cailor de semnalizare STAT5, AKT si MAP kinazei.
Mutatiile activatoare ale exonului 12 conteaza probabil pentru mai putin de 2% din pacientii cu PV, fiind identificate pana in prezent numai la pacienti cu eritrocitoza JAK2V617F(-) si, in majoritatea cazurilor, la cei cu eritrocitoza idiopatica fara leucocitoza si trombocitoza asociate. Aceasta sugereaza faptul ca mutatii activatoare diferite in JAK2 se asociaza cu fenotipuri clinice diferite5;10.
Mutatiile exonului 12 JAK2 sunt suficiente sa produca eritrocitoza in stare heterozigota, rezultand intr-o semnalizare ligand-independenta prin JAK2 mai puternica, iar proteine tinta, cum ar fi Stat5, ERK1 si ERK2 par sa fie mai puternic fosforilate decat in cazul JAK2V617F8;9. Absenta mutatiilor exonului 12 la pacientii cu TE este in acord cu conceptul ca niveluri scazute ale semnalizarii JAK2 favorizeaza trombocitoza, in timp ce semnalizarea mai activa favorizeaza eritrocitoza8.
Pacientii cu mutatii ale exonului 12 JAK2 se prezinta cu eritrocitoza, niveluri scazute ele eritropoietinei serice si cu histologie medulara distinctiva (hiperplazie eritroida fara anomalii morfologice ale megakariocitelor si liniei granulocitare). Ca si in alte MPN, se pot obtine culturi eritroide eritropoietin-independente din celulele sanguine periferice, iar anomaliile citogenetice, splenomegalia sau transformarea mielofibrotica au fost observate la unii pacienti8.
Recomandari pentru mutatia JAK2V617F si interpretarea rezultatelor
- Evaluarea diagnostica initiala a pacientilor cu suspiciune clinica de MPN si BCR-ABL1(-).
- Evaluarea policitemiilor impreuna cu nivelul seric al eritropoietinei.
Genotiparea moleculara face parte din criteriile de diagnostic WHO 2008 pentru MPN, respectiv testarea pentru mutatiile JAK2, MPL si KIT devenind un instrument standard in protocolul de diagnostic al MPN, detectia uneia din aceste mutatii stabilind cu certitudine prezenta unei boli clonale si excluzand granulocitoza, eritrocitoza, trombocitoza sau mielofibroza reactiva11.
In schimb ele nu pot distinge intre diferitele forme de MPN, desi mutatiile exonului 12 JAK2 nu au fost raportate pana in prezent decat in PV si nici un pacient cu PV nu a prezentat o mutatie MPL10. Pentru un diagnostic definitiv rezultatul trebuie corelat cu datele clinice si alte date de laborator, precum si cu modificarile histopatologice caracteristice fiecarui subtip11 (vezi figura 3).
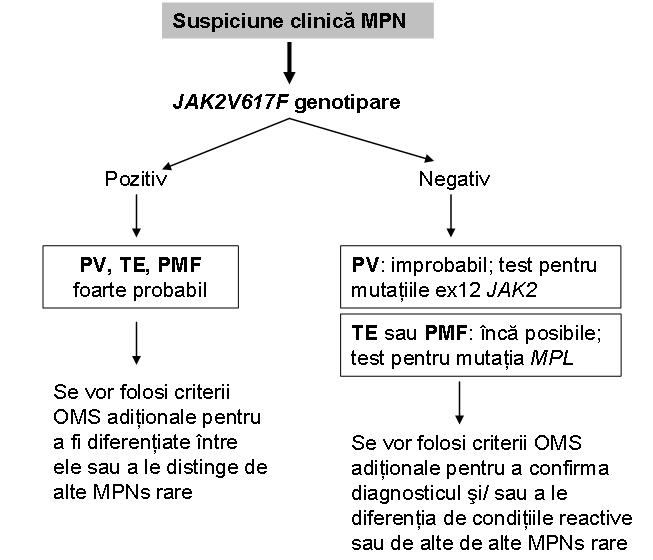
Fig. 3 Algoritm de utilizare a genotiparii JAK2V617F in cazul suspiciunii clinice de MPN.
(Adaptare dupa Vannucchi M A, Guglielmelli P, Tefferi A. „Advance in Understanding and Management of Myeloproliferative Neoplasms”. CA Cancer J Clin 2009; 59: 171-191).
Semnificatia prognostica a detectiei mutatiei JAK2V617F in MPN nu a fost inca clar stabilita.
Un rezultat negativ pentru mutatia JAK2V617F nu exclude prezenta unei MPN sau altei neoplazii. In cazurile rare de PV la care aceasta este absenta poate fi gasita o mutatie a exonului 12 JAK2, iar un procent redus dintre pacientii cu PMF si TE la care lipseste o mutatie JAK2 poate demonstra mutatii activatoare MPL, cum ar fi MPL W515K sau MPL W515L11. Este insa in dezbatere daca diagnosticul de PV poate fi sustinut in absenta unei mutatii JAK210 .
Datorita implicatiilor terapeutice testarea pentru BCR-ABL1 trebuie considerata la pacientii fara mutatii JAK2 sau MPL1.
Specimen recoltat – sange venos4.
Recipient de recoltare – vacutainer ce contine EDTA ca anticoagulant4.
Cantitate recoltata – cat permite vacuumul4.
Cauze de respingere a probei – folosirea heparinei ca anticoagulant4.
Stabilitate proba – 7 zile la 2-8ºC4.
Metoda – secventierea exonului 14. La pacientii negativi pentru mutatia JAK2V617F se va efectua secventierea exonului 12 JAK24.
Limite si interferente
Pentru diagnosticul molecular al mutatiilor exonului 12 este de preferat sa se foloseasca ADN din celulele medulare sau din colonii hematopoietice clonogene decat ADN din leucocitele periferice, deoarece implicarea granulocitara la acesti pacienti este de obicei scazuta8.
Metoda folosita poate sa nu identifice mutatiile la pacientii cu incarcatura clonala mica6.
Bibliografie
1. Beer A P, Green R A. „Pathogenesis and management of essential thrombocythemia”. Hematology 2009(1): 621.
2. George I T. „Pathology of the Myeloproliferative Diseases”. In Wintrobe`s Clinical Hematology, Greer J, Foerster J, Lukens J, Rodgers G, Pareskevas F, Glader B, 11th ed, Lippincott Williams & Wilkins, 2004, 2054-66. 1988.
3. James C. „The JAK2V617F Mutation in Polycythemia Vera and Other Myeloproliferative Disorders: One Mutation for Three Diseases?”. Hematology 2008(1): 69.
4. Laborator Synevo. Referintele specifice tehnologiei de lucru utilizate, 2015. Ref Type: Catalog.
5. Levine R, Gilliland D G. „Myeloproliferative disorders”. Blood 2008; 112(6): 2190-98.
6. Ohyashiki H J, Hisatomi H, Shimizu S, Sugaya M, Ohyashiki K. „Detection of Low Allele Burden of Jak2 Exon 12 MutationsUsing TA-cloning in Patients with Erythrocytosis”. Japanese Journal of Clinical Oncology 2009; 39(8): 509-513.
7. Rane S, Reddy E P. „Janus kinases: components of multiple signaling pathways”. Oncogene 2000; 19(49): 5662-79.
8. Scott M L, et al. „JAK2 Exon 12 Mutations in Polycythemia Vera and Idiopathic Erythrocytosis”. NEJM 2007; 356(5): 459-468.
9. Skoda R. „Thrombocytosis”. Hematology 2009(1): 159.
10. Vannucchi M A, Guglielmelli P, Tefferi A. „Advance in Understanding and Management of Myeloproliferative Neoplasms”. CA Cancer J Clin 2009; 59: 171-191.
11. Vardiman W J, et al. „The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes”. Blood 2009; 114(5): 937-951.
12. Verstovsek S. „Therapeutic potential of Jak2 inhibitors”. Hematology 2009(1): 636.
13. Yamaoka K, Saharinen P, Pesu M, Holt ET V III, Silvennoinen O, O`Shea J J. „The Janus kinases (Jaks)”. Genome Biol. 2004; 5(12): 253.
Produsul a fost adăugat în coș
În plus, ai la dispoziție 30 de zile pentru a veni la recoltare.