- Teste de hematologie
- Teste de biochimie
- Biochimie generală din sânge și urina
- Proteine specifice in ser si urina
- Teste biochimice din lichide de punctie
- Teste biochimice din materii fecale
- Teste biochimice pentru tulburari ereditare de metabolism
- Teste pentru nefrolitiaza
- Vitamine, oligoelemente, stres oxidativ
- Acizi grași
- Transferina carbohidrat deficitara (CDT) marker pentru alcoolism
- Markeri non-invazivi pentru afecţiunile hepatice
- Analiza chimică calculi
- Markeri endocrini
- Markeri tumorali
- Markeri virali
- Markeri cardiaci
- Markeri anemie
- Markeri ososi
- Markeri boli autoimune
- Anticorpi antispermatozoizi
- Autoanticorpi in afectiuni endocrine, cardiace, renale
- Autoanticorpi in afectiuni neurologice
- Autoanticorpi in afectiunile dermatologice
- Autoanticorpi in anemia pernicioasa
- Autoanticorpi in diabetul zaharat
- Markeri pentru afectiuni hepatice si gastrointestinale autoimune
- Markeri pentru afectiuni reumatismale si vasculite
- Markeri pentru monitorizarea evolutiei si tratamentului
- Markeri pentru sindromul antifosfolipidic
- Serologie boli infectioase
- Teste specializate de alergologie si imunologie
- Teste de biologie moleculara
- Teste de citogenetica
- Teste de microbiologie
- Toxicologie
- Citologie cervico-vaginala
- Histopatologie
- Consult genetic
- Genetica medicala

Hemofilia A – analiza inversii IVS1 si IVS2
Preț: 1903.00 lei
Informatii generale
Hemofilia A (hemofilia clasica), cauzata de deficienta activitatii coagulante a FVIII, reprezinta, ca frecventa, cea de-a doua boala hemoragica mostenita dupa boala von Willebrand, cu o incidenta de pana la 1/10000 persoane, cu o prevalenta similara intre diferitele grupe rasiale1;2. Hemofilia A este exemplul clasic de tara recesiva X-linkata, boala fiind cauzata de mutatii heterogene la nivelul genei FVIII (F8) de pe cromozomul Xp286.
Boala se manifesta la barbati care nu prezinta o alela normala; acestia nu transmit anomalia la fiii lor, dar toate fiicele sunt purtatoare ale tarei. Majoritatea femeilor carrier nu sunt afectate datorita prezentei unei alele normale mostenite de la mama, iar ele vor transmite boala la jumatate din fiii lor si statusul de carrier la jumatate din fiice2.
Indivizii afectati prezinta un fenotip variabil in functie de severitatea deficientei FVIII, respectiv expresivitatea defectului genetic, dar in cadrul aceleeasi familii severitatea clinica este constanta, desi alti factori genetici sau de mediu pot modifica intr-o anumita masura severitatea clinica1;2. Astfel, pacientii cu hemofilie A severa (nivel de FVIII <1%) sunt diagnosticati in primul an de viata, sangerarile musculare profunde si hemartrozele spontane reprezentand simptomele cele mai frecvente. Indivizii cu hemofilie A moderat severa (FVIII = 2-5%) prezinta de obicei sangerari prelungite sau intarziate dupa traume minore si sunt diagnosticati in general inainte de varsta de 5-6 ani. Indivizii cu hemofilie A usoara (FVIII = 5-30%) prezinta sangerari anormale dupa interventii chirurgicale, extractii dentare sau traume majore, acestia fiind de obicei diagnosticati mai tarziu in timpul vietii1;5. Proportia cazurilor severe, moderate, usoare este de 50%, 10%, respectiv 40%6. Aproximativ 10% din femeile carrier au nivelul FVIII <35% si sunt simptomatice, simptomele fiind de obicei usoare, variatiile in nivelul FVIII fiind atribuite lionizarii (inactivarea cromozomului X normal in cursul embriogenezei)1;5;6. Alte cauze ale hemofiliei A la femei sunt: copilul de sex feminin mosteneste doi cromozomi X anormali de la tatal hemofilic si mama carrier; anumite boli cromozomiale pot duce la aparitia unui genotip hemizigot si induc hemofilia prin absenta unei alele normale: mozaicism 45XX/45X, cariotip 46XY, deletia cromozomului X (genotip XO, sindrom Turner). In cazuri rare bolnavi nehemofilici, cu boli autoimune (lupus eritematos sistemic, artrita reumatoida) sau dupa administrare de medicamente (peniciline, fenitoin) sau cu boli limfoproliferative, pot dezvolta autoanticorpi anti-FVIII care determina aparitia hemofiliei A dobandite.
Comostenirea mutatiei factorului V Leiden sau a altor factori de risc protrombotic poate explica variabilitatea fenotipului intre pacienti cu acelasi defect molecular al FVIII2.
Rosendaal si altii (1990) au prezentat dovezi ca mortalitatea datorata bolii ischemice cardiace este mai mica la hemofilici decat in populatia generala6.
Gena FVIII are 186 bp (perechi de baze), cu 26 exoni si 25 introni, continand insule CpG, care predispun gena la mutatii si codifica un ARNm de 9 kb, aproximativ 5% din ADN fiind localizat in exoni, iar 95% reprezentand “non-coding” ADN. FVIII este exprimat cu un peptid semnal de 19 aminoacizi, proteina matura are 2332 reziduuri, structura domeniilor dinspre capatul amino-terminal fiind “A1-A2-B-A3-C1-C2”, omologa FV al coagularii. Este sintetizat in principal in ficat si circula in plasma sub forma inactiva fiind stabilizat prin legare de FVW. O data activat de catre trombina, este eliberat de FVW si se leaga la suprafata membranara fosfolipidica.
Diagnosticul de hemofilie A este stabilit la indivizi cu nivel scazut al activitatii coagulante a FVIII in prezenta unui nivel normal de FVW1;4.
Recomandari pentru testarea genetica in hemofilia A1;5
- Testarea genetica a unui individ afectat pentru detectia mutatiei specifice a FVIII intr-o familie, in vederea obtinerii de informatii necesare consilierii genetice a membrilor familiei cu risc crescut.
- Diagnosticul diferential cu boala von Willebrand, in special tipul 3 (care clinic si biochimic este similara hemofiliei A usoare), constand in testarea genetica a genei FVIII, testarea genetica a genei FVW sau masurarea activitatii FVW de legare a FVIII (cand aceasta este disponibila).
- La indivizii care reprezinta singurul caz din familie identificarea mutatiei specifice poate ajuta la predictia fenotipului clinic si stabilirea riscului de a dezvolta inhibitori ai FVIII.
- Testarea pentru carrier a rudelor cu risc (necesita identificarea mutatiei cauzatoare in familie). Se recomanda ca statusul de carrier sa fie stabilit inaintea sarcinii sau cat mai devreme in sarcina.
- Diagnosticul prenatal si preimplantare pentru sarcinile cu risc (necesita identificarea mutatiei cauzatoare in familie). Procedura uzuala consta in determinarea sexului fetal prin analiza cromozomilor in celulele fetale obtinute din vilozitatile coriale la 10-12 saptamani de sarcina sau prin amniocenteza la 15-18 saptamani, iar daca carioptipul este 46XY, ADN-ul extras din celulele fetale poate fi analizat pentru prezenta mutatiei sau pentru markerii informativi. Daca mutatia nu este cunoscuta, iar linkajul nu este informativ, diagnosticul prenatal este posibil prin masurarea activitatii coagulante a FVIII in sangele obtinut prin punctia percutana a cordonului ombilical la 18-21 saptamani de sarcina (risc 1-6% de moarte fetala).
Specimen recoltat – sange venos3.
Recipient de recoltare – vacutainer ce contine EDTA ca anticoagulant3.
Cantitate recoltata – cat permite vacuumul3.
Cauze de respingere a probei – folosirea heparinei ca anticoagulant3.
Stabilitate proba – 7 zile la 2-8ºC3.
Metoda – sunt disponibile doua variante de testare:
-analiza inversiilor IVS1 si IVS22 prin reactie de polimerizare in lant (PCR) este prima recomandata la pacientii cu hemofilie A severa;
-hemofilia A panel extins: secventierea genei F8 si analiza deletiilor/duplicatiilor prin MLPA, la pacientii cu hemofilie A severa la care nu se depisteaza una din cele doua inversii, precum si la pacientii cu hemofilie A usoara sau moderata1,3.
Interpretarea rezultatelor
Aproximativ 98% din pacientii cu hemofilie A prezinta mutatii la nivelul genei F8, fiind raportate peste 1000 mutatii, incluzand deletii, insertii, mutatiile punctiforme ale dinucleotidelor CpG fiind in mod special comune. Deletii largi (>50 nucleotide) apar la aproximativ 5% din pacientii cu hemofilie A. Aproximativ 40% din cazurile de hemofilie A severa rezulta dintr-o inversiune majora a varfului bratului lung al cromozomului X, unul dintre punctele sale de ruptura (breakpoints) fiind situate la nivelul intronului 22. O alta inversiune comuna, la nivelul intronului 1, apare in 2-3% din deficientele severe de FVIII2;5.
Gena F8A reprezinta o gena situata in interiorul genei FVIII, continuta in intregime in intronul 22, lipsita de introni si fiind transcrisa in directie inversa fata de FVIII. Cromozomul X contine 3 copii ale F8A si regiunilor sale adiacente, una in intronul 22 si doua telomeric si la ~500kb in amonte de situsul de start al transcriptiei FVIII. Intronul 22 este neobisnuit din multe puncte de vedere. Este cel mai mare intron al genei FVIII (32 kb) si, de asemenea, contine o insula CpG localizata ~10kb in aval de exonul 22, aceasta parand a servi ca promoter bidirectional pentru F8A si F8B (gena F8B este de asemenea localizata in intronul 22 si este transcrisa in directie inversa fata de FVIII). Multe din mutatiile care rezulta in hemofilia A severa sunt bazate pe recombinarea dintre secventele omologe F8A din intronul 22 si din amonte de gena FVIII, ducand la inversiunea si translocarea exonilor 1-22 fata de exonii 23-26. Acest defect isi are originea in principal in celulele germinale masculine, in timpul diviziunii celulare meiotice, cand lipsa unei formatiuni bivalente faciliteaza “indoirea” (“flipping”) capatului telomeric al cromozomului X unic4,6.
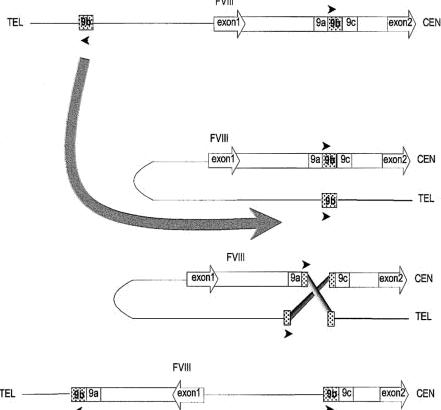
Factorul VIII cu inversia in intronul 1 din cromozomul X.
Intronul 1, flancat de exoni, orientati in directia transcriptiei, continand secventa repetitiva 9b flancata de secventele unice 9a si 9c; in urma recombinarii intre cele doua secvente 9b are loc inversia. CEN-centromeric; TEL-telomeric. Richard D. Bagnall, Naushin Waseem, Peter M. Green and Francesco Giannelli, Blood, 2002, 99: 168-174.
Rata de detectie a mutatiilor la indivizii cu hemofilie A care nu prezinta una din cele doua inversiuni comune variaza intre 75-98%. In hemofilia A severa alterari grosiere ale genei (deletii sau insertii largi, mutatii ale cadrului de citire sau ale jonctiunii de “splicing’, mutatii nonsense si missense) conteaza pentru aproximativ 50% din mutatiile detectate1.
In schimb la pacientii cu hemofilie A moderata sau usoara cel mai frecvent sunt detectate mutatii punctiforme missens la nivelul exonilor care codifica domeniile A sau C1;2 , aproximativ jumatate din mutatii fiind grupate la nivelul domeniului A26.
Produsii genei anormale variaza intre absenta unei proteine detectabile (majoritatea cazurilor de hemofilie A severa) la niveluri normale ale unei proteine disfunctionale (5% din cazuri), restul pacientilor (45%) avand niveluri concordant scazute ale antigenului si activitatii coagulante a FVIII. Anumite mutatii cum ar fi codoni terminali prematuri, inversii sau deletii largi ale genei se asociaza cu risc crescut de aparitie a inhibitorilor1;6.
Exista elemente ale istoricului familial predictoare pentru statusul de carrier al unei femei:
-o femeie care are un fiu afectat si o alta ruda afectata pe linie materna este carrier obligatoriu;
-daca o femeie are mai mult de un fiu afectat si mutatia nu poate fi detectata la nivelul ADN-ului sau, atunci ea prezinta mozaicism al liniei germinale (rar).
Hemofilia A rezulta si ca urmare a unei rate crescute de mutatii spontane, favorizate de dimensiunile mari ale genei (186kb) si de prezenta de puncte fierbinti (“hot spots”), cum ar fi dinucleotidele CpG, aceste mutatii aparand de cel putin 3 ori mai frecvent la barbati decat la femei. In fapt aproximativ o treime din hemofilici nu prezinta istoric familial de sangerari anormale2. Daca un pacient este un caz simplex (fara istoric familial de hemofilie), exista cateva posibilitati in ceea ce priveste statusul de carrier al mamei sale:
-mama nu este carrier si pacientul prezinta o mutatie de novo;
-mama este carrier de novo, mutatia aparand ca o mutatie a liniei germinale (prezenta in toate celulele si detectabila la nivelul ADN-ului), ca o mutatie somatica (prezenta in unele celule si care poate sa nu fie detectabila la nivelul ADN-ului) sau mozaicism al liniei germinale (mutatia este prezenta in unele celule germinale, dar nu este detectabila in ADN-ul din leucocite);
-mama este carrier si a mostenit mutatia cauzatoare de la mama, care are o mutatie de novo sau de la tatal asimptomatic care este mozaic pentru mutatie;
-mama este carrier pentru o mutatie aparuta in generatiile anterioare, care a trecut prin familie, fiind asimptomatica la femeile carrier.
Per ansamblu mama are 80% sanse sa fie carrier, dar sansele sunt 98% daca pacientul prezinta o forma severa de boala asociata cu inversiunea intronului 221.
Mozaicismul poate reprezenta un eveniment destul de comun in hemofilia A; astfel stabilirea riscului in cadrul consilierii genetice trebuie sa ia in considerare posibilitatea mozaicismului somatic in familiile cu mutatii aparent de novo, in special in cazul mutatiilor punctiforme5;6.
Au fast raportate cazuri de hemofilie A cu transmitere autozomal dominanta care trebuie diferentiate de tipul 2N de boala vonWillebrand2.
Limite si interferente
Cand testarea pentru carrier este efectuata fara identificarea anterioara a mutatiei in familie, obtinerea unui rezultat negativ la o ruda cu risc nu este informativa.
Masurarea activitatii coagulante a FVIII nu este suficienta pentru determinarea statusului de carrier, ea fiind doar sugestiva daca este scazuta1.
Bibliografie
1. Brower C, Thompson A. „Hemophilia A”. GeneReviews, NCBI Bookshelf. Ref Type: Internet Communication.
2. Friedman K, Rodgers G. „Inherited Coagulation Disorders”. In Wintrobe`s Clinical Hematology, Greer J, Foerster J, Lukens J, Rodgers G, Pareskevas F, Glader B, 11th ed, Lippincott Williams & Wilkins, 2004, 1379-82.
3. Laborator Synevo. Referintele specifice tehnologiei de lucru utilizate, 2010. Ref Type: Catalog.
4. Maclean R, Makris M. „Hemophilia A and B”. In Practical Hemostasis and Thrombosis, O’Shaughnessy D, Makris M, Lillicrap D eds, Blackwell Publishing, 2008, 41-43.
5. Mayo Clinic, Mayo Medical Laboratories. Reference Laboratory Services for Health Care Organizations. “Hemophilia A, Molecular Analysis for Inversion, Diagnosis and Carrier Detection”. www.mayomedicallaboratories.com. 2010. Ref Type: Internet Communication.
6. Online Mendelian Inheritance in Man, McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine. „Hemophilia A”. Ref Type: Internet Communication.
Produsul a fost adăugat în coș
În plus, ai la dispoziție 30 de zile pentru a veni la recoltare.