- Teste de hematologie
- Teste de biochimie
- Biochimie generală din sânge și urina
- Proteine specifice in ser si urina
- Teste biochimice din lichide de punctie
- Teste biochimice din materii fecale
- Teste biochimice pentru tulburari ereditare de metabolism
- Teste pentru nefrolitiaza
- Vitamine, oligoelemente, stres oxidativ
- Acizi grași
- Transferina carbohidrat deficitara (CDT) marker pentru alcoolism
- Markeri non-invazivi pentru afecţiunile hepatice
- Analiza chimică calculi
- Markeri endocrini
- Markeri tumorali
- Markeri virali
- Markeri cardiaci
- Markeri anemie
- Markeri ososi
- Markeri boli autoimune
- Anticorpi antispermatozoizi
- Autoanticorpi in afectiuni endocrine, cardiace, renale
- Autoanticorpi in afectiuni neurologice
- Autoanticorpi in afectiunile dermatologice
- Autoanticorpi in anemia pernicioasa
- Autoanticorpi in diabetul zaharat
- Markeri pentru afectiuni hepatice si gastrointestinale autoimune
- Markeri pentru afectiuni reumatismale si vasculite
- Markeri pentru monitorizarea evolutiei si tratamentului
- Markeri pentru sindromul antifosfolipidic
- Serologie boli infectioase
- Teste specializate de alergologie si imunologie
- Teste de biologie moleculara
- Teste de citogenetica
- Teste de microbiologie
- Toxicologie
- Citologie cervico-vaginala
- Histopatologie
- Consult genetic
- Genetica medicala

Fibroza chistică – panel extins (secvențiere completă CFTR)
Preț: 4650.00 lei
Informaţii generale şi recomandări pentru efectuarea testului genetic
Fibroza chistica sau mucoviscidoza este cea mai frecventa afectiune monogenica, avand o incidenta de 1:2500-3500 nou-nascuti in randul populatiei de origine caucaziana. Se transmite autozomal recesiv si se caracterizeaza prin pleiomorfism clinic, cu evolutie cronica, avand o durata medie de supravietuire de 37 ani, varsta ce este mai mare la barbati decat la femei.
|
Rasa sau grup etnic |
Frecventa purtatorilor (heterozigotilor) |
Rata de detectie a mutatiilor |
|
Americani de origine africana |
1/65 |
77% |
|
Evreii Ashkenazi |
1/25 |
99% |
|
Americani de origine asiatica |
1/90 |
54% |
|
Europeni |
1/25 |
80% |
|
Est europeni |
1/25 |
75% |
|
Romani |
1/30 |
65% |
|
Canadieni de origine franceza |
1/25 |
91% |
|
Americani de origine hispanica |
1/46 |
81% |
|
Nord europeni |
1/25 |
91% |
|
Sud europeni (italiani) |
1/25 |
77%5 |
Boala este cauzata de mutatii la nivelul unei genei situate pe bratul lung al cromozomului 7 (7q31), care codifica o proteina transmembranara, CFTR (Cystic Fibrosis Transmembrane Conductance Regulator), ce face parte din familia proteinelor cu activitate ATP-azica si functioneaza ca un canal de clor la nivelul polului apical al membranei celulelor epiteliale. In plus, proteina este implicata si in reglarea canalelor de sodiu, intervine in transportul HCO3– prin membranele celulelor epiteliale si poate actiona ca un canal pentru alte proteine, cum ar fi glutationul. Studii proteomice recente au demonstrat ca CFTR interactioneaza cu multe proteine intracelulare insa relevanta fiziopatologica a acestor interactiuni nu a fost inca pe deplin elucidata1.
Dupa depistarea in 1989 a defectului genetic de la nivelul genei implicate in fibroza chistica, s-a crezut ca un numar limitat de mutatii cauzeaza aceasta boala, insa pana in prezent au fost descrise mai mult de 1500 mutatii diferite. Aproape toate sunt mutatii punctiforme sau deletii mici (de la 1 la 84 bp). Cu toate acestea este important sa se inteleaga ca majoritatea sunt rare, iar consecintele functionale ale multora dintre ele sunt greu de inteles. De fapt mai putin de 10 mutatii (evidentiate in tabelul 2), apar cu o frecventa mai mare de 1%, in timp ce cea mai frecventa mutatie la nivel mondial, caracterizata prin deletia fenilalaninei in pozitia 508 (ΔF508 – Phe508del) (deletia a 3 perechi de baze de la nivelul exonului 10), se gaseste la aproximativ 30-80% dintre pacientii cu mucoviscidoza, in functie de grupul etnic afectat. In tabelul 1 sunt enumerate 25 dintre mutatiile testate in laboratorul nostru pentru stabilirea diagnosticului de fibroza chistica sau testarea purtatorilor1;3.
Mutatiile genei CFTR pot fi grupate in 6 clase diferite, impartite in raport cu consecintele lor functionale la nivel celular (figura 1):
-clasa I: proteina nu este sintetizata;
-clasa II: CFTR este insuficient prelucrata la nivelul aparatului Golgi;
-clasa III: proteina nu este functionala;
-clasa IV: CFTR prezinta conductanta anormala;
-clasa V: CFTR prezinta un defect partial de sinteza;
-clasa VI: CFTR este degradata accelerat1.
Mutatiile din clasele I, II, III sunt mai frecvente si se asociaza cu insuficienta pancreatica, in timp ce mutatiile din clasele IV, V si VI sunt mai rare, iar pacientii nu prezinta manifestari pancreatice1.

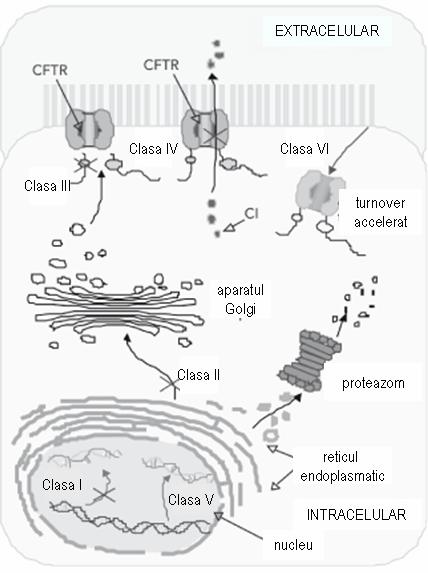
Figura 1 adaptare dupa Roberta Rodrigues; Carmen S. Gabetta, Karla P. Pedro, Fabio Valdetaro, Maria I. M. Fernandes, Patrícia K. R. Magalhães, José N. Januário, Léa M. Z. MacielI, Cystic fibrosis and neonatal screening, Cad. Saúde Pública vol.24 suppl.4, 2008.
Tabelul 1
|
3120+1G>A |
A455E |
G85E |
R334W |
1717-1G>A |
|
3659delC |
ΔF508 |
R347P |
1898+1G>A |
3849+10kbC>T |
|
ΔI507 |
N1303K |
R553X |
2184delA |
621+1G>T |
|
G542X |
R1162X |
R560T |
2789+5G>A |
711+1G>T |
|
G551D |
R117H |
W1282X |
1078delT |
I148T6 |
Tabelul 2
|
Mutatii |
Frecventa |
Clasa |
Fenotipul |
|
ΔF508 |
66.0% |
II |
Clasic |
|
G542X |
2.4% |
I |
|
|
G551D |
1.6% |
III |
|
|
N1303Lys |
1.3% |
II |
|
|
W1282X |
1.2% |
I |
|
|
R553X |
0.7% |
I |
|
|
621+1G>T |
0.7% |
I |
|
|
1717-1G>A |
0.6% |
I |
|
|
R117H |
0.3% |
IV |
Non-clasic |
|
R1162X |
0.3% |
Nedeterminata * |
Clasic6 |
* Transcriptia se realizeaza in conditii normale, dar proteina este trunchiata, probabil incorect impachetata, de aceea cel mai probabil face parte din clasa II.
Gena CFTR contine aproximativ 250-280 kilobaze, fiind alcatuita din 27 exoni. Aceasta codifica o glicoproteina constituita din 1480 aminoacizi, ce formeaza cinci domenii: 2 domenii transmembranare, fiecare cu 6 deschideri in α-helix, 2 domenii nucleotidice (NBD) in citoplasma, interconectate la regiunile transmembranare si un domeniu reglator (R), care leaga intre ele domeniile transmembranare. Canalul ionic se deschide doar atunci cand regiunea reglatoare a fost fosforilata de catre proteinkinaza A (PKA), iar domeniul nuceotidic leaga ATP.

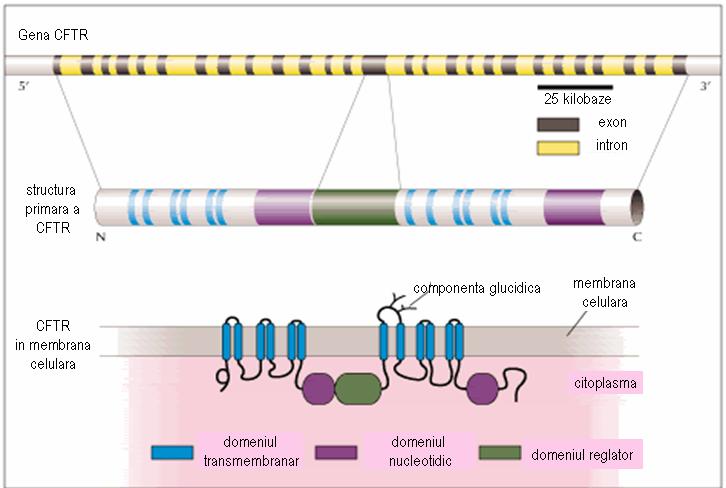
Figura 2 Adaptare dupa http://www.chromosome7.htmlplanet.com/custom4.html
Consecinta anomaliilor genetice este reprezentata de absenta sau functionarea inadecvata a canalelor de clor la nivel celular, ceea ce se traduce prin alterarea transportului clorurilor in glandele mucoase si seroase de la nivelul majoritatii organelor. Aceste secretii vor avea un continut scazut de apa, vor fi vascoase, aderente la epiteliile ductelor excretoare si greu de eliminat spre exterior. Acumularea acestora produce in timp alterarea functiilor si distructia diferitelor organe (plamani, pancreas, ficat, intestin, organe de reproducere). La nivelul pielii apare o secretie sudorala cu concentratie ridicata de sare.
Mucoviscidoza variaza ca severitate in functie de mutatiile CFTR si factorii de mediu si se prezinta sub mai multe forme, unele care determina moarte precoce a copiilor, ca urmare a unei boli pulmonare obstructive progresive cu bronsiectazii, altele caracterizate prin insuficienta pancreatica si boala pulmonara obstructiva progresiva in timpul adolescentei cu cresterea frecventei de spitalizare la maturitate, iar altele manifestate prin sinuzite si bronsite recurente sau infertilitate la barbatii tineri.
Prezentarea clinica, varsta la diagnostic, severitatea simptomelor si rata de progresie a bolii in organele implicate variaza pe scara larga1;2;6.
Manifestari pulmonare. In utero, la nastere si imediat dupa aceea copiii cu fibroza chistica prezinta histologie normala a plamanilor (cu exceptia ductelor glandelor submucoase din caile respiratorii care sunt dilatate). La scurt timp insa, apar modificarile pulmonare, ce se caracterizeaza prin obstructia cailor aeriene periferice ca urmare a acumularii de secretii. Persoanele afectate dezvolta ulterior un sindrom inflamator la nivelul tractului respirator inferior si apoi infectii cronice endobronsice. Deoarece elastaza (NE) eliberata de neutrofile la locul inflamatiei determina clivajul receptorului CR1, a componentei C3b a complementului si a imunoglobulinei G (IgG), bacteriile nu mai pot fi opsonizate si distruse fiind astfel favorizata persistenta acestora. Alterarea apararii antiinfectioase duce la aparitia bronsitelor si bronsiolitelor bacteriene (cel mai frecvent cu Staphylococcus aureus si Pseudomonas aeruginosa, dar si Aspergillus fumigatus cu dezvoltarea de micetoame), cu infiltratie neutrofilica intensa si obstructia tractului respirator. Prezenta elastazei determina de asemenea cresterea la nivelul celulelor epiteliale a productiei de IL-8, care este chemoatractant pentru neutrofile, distruge elastina si actioneaza ca secretagog, contribuind la mentinerea inflamatiei si infectiei si producand astfel leziuni structurale si tulburari ale schimbului de gaze2.
Simptomele respiratorii pot incepe din prima luna de viata cu tuse, tahipnee sau wheezing, unii copii putand prezenta chiar detresa respiratorie severa asociata cu bronsiolita.
Deoarece polipii nazali apar la 10-32% dintre pacientii cu fibroza chistica, prezenta acestora reprezinta o indicatie pentru efectuarea testului sudorii. Majoritatea copiilor de peste 8 luni (90%) prezinta sinuzita recurenta refractara la terapia cu antibiotice.
La adulti manifestarile mucoviscidozei se caracterizeaza prin tuse cronica (la inceput uscata, intermitenta, concordanta cu episoadele infectioase, ulterior se prelungeste in timp, devine paroxistica, cu exacerbari nocturne si in special dimineata la trezire, apoi productiva), expectoratie intermitenta cu cresterea productiei de sputa asociata cu modificari ale culorii acesteia, febra usoara, dispnee de efort, pneumonie, bronsiolita recurenta, atelectazie, hiperreactivitatea cailor respiratorii cu aparitia astmului refractar si lipsa raspunsului la tratamentul cu agenti β-adrenergici, bronsiectazii, hemoptizie, pneumotorax si retractii suprasternale si intercostale. Pe masura ce boala pulmonara progreseaza, ca urmare a infectiilor cronice, apar leziuni structurale importante ale cailor respiratorii (chisturi sau abcese) insotite de fibroza parenchimului pulmonar adiacent si aparitia obstructiei cu hiperinflatie si blocarea gazelor. Hipoxemia apare initial in timpul somnului si efortului fizic, iar hipercapnia se dezvolta tardiv si este un element de prognostic negativ. De asemenea aparitia hipertensiunii pulmonare si dezvoltarea cordului pulmonar o data cu boala parenchimatoasa avansata reprezinta un semn de prognostic nefavorabil, care este asociat cu o supravietuire de aproximativ 8 luni.
Un studiu recent a raportat ca expunerea pasiva la fumul de tigara afecteaza negativ functia pulmonara la persoanele cu fibroza chistica.
In ciuda tuturor interventiilor terapeutice bolile pulmonare raman cauza majora de morbiditate si mortalitate in fibroza chistica2;3;5;6.
Manifestari gastrointestinale. Mutatiile de la nivelul genei CFTR determina aparitia unei secretii alterate de clorura si apa la nivel intestinal, ce poate conduce la aparitia ileusului meconial la nastere (mai frecvent la homozigotii ΔF508) sau a sindromului de obstructie intestinala distala mai tarziu in viata.
Ileusul meconial apare la 15-20% din nou-nascutii diagnosticati cu mucoviscidoza, putand fi principala manifestare in perioada neonatala. Sindromul de impactare meconiala este determinat de acumularea de masa mucofecaloida lipicioasa in ileonul terminal si cec, aderenta la peretele intestinal, ce uneori se poate calcifica.
Atrezia jejunala asociata frecvent cu volvulus si peritonita meconiala poate fi de asemenea o forma de prezentare a fibrozei chistice.
Diagnosticul de mucoviscidoza poate fi suspectat inca din viata intrauterina datorita evidentelor ultrasonografice sugestive pentru obstructie intestinala, cum ar fi aspectul hiperecogen al intestinului fetal.
Cauza sindromului de obstructie intestinala distala este neclara, putandu-se asocia cu deshidratarea, febra, reducerea suplimentelor enzimatice, boala hepatica sau utilizarea medicamentelor cu efect antiperistaltic (opiacee, anticolinergice). Clinic se caracterizeaza prin crampe recurente, prezenta unei mase palpabile la nivelul abdomenului si semne de obstructie intestinala completa sau partiala. 50% dintre acesti pacienti sufera complicatii cum ar fi peritonita, volvulus, atrezie, necroza, perforatie sau formarea unui pseudochist.
Impactarea mucoida a apendicelui poate fi observata ca o masa palpabila asimptomatica in cadranul abdominal inferior.
Ca urmare a cresterii presiunii intraabdominale secundare tusei acesti pacienti dezvolta frecvent prolaps rectal.
Colonopatia fibrozanta este o entitate caracterizata prin strictura colonului ascendent, ce a fost descrisa initial in 1994 si este caracteristica fibrozei chistice. Dozele mari de enzime pancreatice, in special lipaze au fost incriminate in etiologia bolii. Simptomele initiale sunt similare cu cele din sindromul obstructiv, iar diagnosticul se stabileste in urma efectuarii unui tranzit baritat.
Incidenta tumorilor gastrointestinale (esofagiene, gastrice, intestinale, hepatice, biliare, pancreatice si retroperitoneale) este in crestere la pacientii cu fibroza chistica, colonul fiind cea mai frecventa localizare a cancerului.
Insuficienta pancreatica exocrina (cauzata de obstructia ductelor pancreatice cu secretii, aparitia autodigestiei enzimatice si in final a fibrozei interstitiale) insotita de sindrom de malabsorbtie apare inca de la nastere la marea majoritate a persoanelor diagnosticate cu FC, cu exceptia celor care au anumite mutatii CFTR si nu necesita administrarea de enzime pancreatice. Acestia risca totusi sa dezvolte pancreatita acuta sau recurenta.
Manifestarile clinice sunt steatoreea, distensia abdominala, durerile abdominale, flatulenta, retardul de crestere (determinat de sindromul de malabsorbtie si anemia hemolitica), defectele de coagulare sau eruptiile cutanate asociate cu deficientele de vitamine liposolubile (A, D, E si K) si zinc. La acesti pacienti se recomanda administrarea de enzime pancreatice. Persoanele cu FC si functie pancreatica nomala au o evolutie clinica mai blanda cu rata medie de supravietuire mai mare (56 ani) fata de cei cu insuficienta pancreatica.
La copii deficitul de vitamine, electroliti si proteine se manifesta prin: fontanele proeminente; anemie hemolitica prin deficitul de vitamina E; complicatii hemoragice ce pot rezulta prin deficitul de vitamina K datorat insuficientei hepatice sau malabsorbtiei; hipoproteinemie si edeme; hepatomegalie insotita de cresterea valorilor enzimelor hepatice; acrodermatita enteropatica, constipatie, deshidratare hiponatremica/hipocloremica si piele sarata, alcaloza metabolica hipokaliemica secundara pierderii cronice de sare.
Scaderea tolerantei la glucoza este frecventa la bolnavii cu mucoviscidoza (in medie 40%). Diabetul zaharat asociat cu fibroza chistica (DZAMV) se manifesta in adolescenta (prin cetoacidoza si cetonurie), fiind diagnosticat la 7% din pacientii cu varsta intre 11 si 17 ani, cu cresterea prevalentei la maturitate. Este mai frecvent la homozigotii ΔF508 si la sexul feminin. DZAMV reprezinta o entitate distincta fata de diabetul de tip 1 sau 2, dar are caracteristici comune cu ambele tipuri. Etiologia este o combinatie intre secretia redusa de insulina (secundara fibrozei pancreatice si numarului redus de celule insulare) si rezistenta periferica la insulina. Aparitia acestui tip de diabet se asociaza cu un declin al functiei pulmonare, cu o stare nutritionala mai precara si cu supravietuire mai scurta.
Odata cu cresterea perioadei de supravietuire la acesti pacienti, bolile hepatobiliare asociate mucoviscidozei au devenit o complicatie serioasa si frecventa ce pot afecta calitatea vietii pacientilor. Implicarea hepatobiliara devine clinic aparenta dupa primii 10 ani de viata.
Absenta proteinei CFTR functionala in celulele epiteliale care tapeteaza ductele biliare determina formarea unei secretii vascoase, care obstrueaza canaliculele. Daca acest proces este extins apare ciroza obstructiva, ce se poate complica cu varice esofagiene, splenomegalie si hipersplenism.
Nou-nascutii cu fibroza chistica pot prezenta icter obstructiv prelungit prin staza biliara intra si extrahepatica.
Calculii biliari au prevalenta mai mare la pacientii cu mucoviscidoza (15% dintre adultii tineri cu fibroza chistica) decat la subiectii normali.
Boala hepatica este a doua cauza de mortalitate dupa afectiunile pulmonare in fibroza chistica2;3;5;6.
Fertilitate. Mai mult de 95% dintre barbatii cu FC sunt infertili, ca urmare a absentei congenitale bilaterale a ductelor deferente si mai rar prin azoospermie obstructiva. Corpul, coada epididimului si veziculele seminale poate fi anormal dilatate sau chiar absente. Doar 1% dintre pacientii cu mucoviscidoza sunt fertili, acestia prezentand forme usoare de boala. Spermatogeneza este normala la acesti barbati.
Femeile cu FC sunt fertile, desi cateva (20%) pot prezenta un mucus cervical anormal care contribuie la infertilitate.
Deoarece supravietuirea indivizilor cu FC s-a imbunatatit considerabil in ultimele decenii, sarcina la femeile cu FC a devenit o problema importanta. Exista studii care afirma ca sarcina decurge bine la aceste pacientele, dar si studii care atesta aparitia unor complicatii.
Factorii de predictie negativi, atat pentru mama cat si pentru fat, in cazul unei sarcini sunt capacitatea vitala fortata (CVF) mai mica de 50% din valoarea prezisa si starea nutritionala proasta. De fapt o valoare a CVF mai mica de 50% din valoarea prezisa reprezinta o contraindicatie absoluta a sarcinii.
Cu toate acestea tratamentul adecvat al afectiunilor pulmonare, managementul agresiv al infectiilor cu o mare varietate de antibiotice si imbunatatirea nutritiei au facut ca sarcina sa fie bine tolerata, in special la femeile cu forme usoare sau moderate. La acestea factorii de risc pentru deteriorarea sanatatii si deces precoce dupa sarcina sunt aceleasi ca si pentru populatia adulta non-gravida. Intr-un studiu recent, Goss et al a luat in considerare valoarea VEF (volumul expirator fortat), greutatea, inaltimea si rata de exacerbare pulmonara pe an si a constatat ca sarcina nu a fost asociata cu un risc crescut de deces. De fapt sarcina nu pare sa prezinte riscuri suplimentare nici pentru subgrupurile de femei cu diabet zaharat sau cu valoare FEV mai mica de 40% din valoarea prezisa. Factorii predictori importanti pe parcursul sarcinii sunt severitatea insuficientei pulmonare materne si starea de nutritie, in sensul ca deteriorarea acestora pot precipita nastere prematura. In ciuda tuturor eforturilor rata de nou-nascuti vii la femeile cu mucoviscidoza cu varste cuprinse intre 13 si 45 ani este de 1.9%.
Riscul de aparitie a anomalilor congenitale la fat nu este crescut, iar alaptatul nu reprezinta o contraindicatie2;3;6.
Manifestari osteoarticulare. Osteoartropatia hipertrofica afecteaza peste 6% dintre bolnavi, fiind mai rara la copiii mici si mai frecventa la sexul masculin (2:1). Este un sindrom caracterizat prin proliferare anormala a tesutului osos la nivelul extremitatilor distale ale oaselor lungi, cu aparitia hipocratismului digital, durerii sau tumefactiei articulare, de obicei simetrica, si a periostitei.
Artropatia in fibroza chistica apare la 10% dintre bolnavi, putandu-se manifesta in orice moment evolutiv. Etiologic sunt implicate mecanisme imunologice probabil legate de infectia cronica cu Pseudomonas aeruginosa. Debutul este brusc cu durere, tumefactie si uneori se poate asocia cu leziuni tegumentare cum ar fi rash-ul maculopapular, eritemul nodos si ocazional purpura. Poate fi mono- sau poliarticulara, afectand articulatiile mari sau mici, cele mai frecvente fiind genunchiul, glezna, articulatia mainii, cotului sau umarului.
Osteopenia sau osteoporoza sunt manifestari frecvente ale pacientii cu FC si sunt asociate cu aparitia fracturilor si a cifozei3.
Clasic diagnosticul fibrozei chistice se stabileste pe baza elementelor clinico-anamnestice caracteristice si este apoi confirmat prin testul sudorii sau analiza moleculara.
La 70% dintre pacienti diagnosticul este stabilit inaintea varstei de 1 an, de obicei in primele luni de viata. Totusi exista bolnavi la care diagnosticul este confirmat abia dupa varsta de 10 ani.
Diagnosticul de FC poate fi stabilit la persoanele suspicionate daca prezinta:
1. una sau mai multe caracteristici fenotipice ale FC;
si
2. dovada anomaliei functiei CFTR:
-prezenta mutatiilor cauzatoare de boala in gena CFTR;
sau
-2 valori anormale ale clorurilor in sudoare la iontoforeza cantitativa pilocarpinica (> 60 mEq/L);
sau
-valori specifice ale diferentei de potential nazal.
Testul sudorii ramane gold standard-ul in diagnosticarea bolii si evalueaza concentratia de ioni de clor si sodiu din transpiratie. Valorile normale ale electrolitilor in sudoare se situeaza <40mMol/L; valorile pozitive sunt la copii >60 mMol/L, iar la adolescenti si adulti tineri >70 mMol/lL; valorile echivoce: intre 40-60 mMol/L se repeta obligatoriu si se interpreteza in context clinic. O concentratie de clorura de mai mare de 60 mMol/L in sudoare, la doua determinari diferite stabileste diagnosticul de boala.
Rezultatele fals pozitive pot fi asociate cu sindromul Hurler, iar cele fals negative pot sa apara in cazul pierderilor acute de sare. In cazul in care FC este suspectata la un individ cu hiponatremie si hipocloremie, testul sudorii ar trebui amanat pana la restabilirea unui echilibrul electrolitic3;6.
In urmatoarele situatii speciale testarea genetica este testul initial de diagnostic:
-diagnosticarea in utero a fetilor cu risc ridicat (in 2002, 4% din persoanele nou diagnosticate au fost identificate cu ajutorul diagnosticului prenatal);
-testarea prenatala a fetilor cu risc scazut, dar cu imagini ecografice sugestive pentru boala;
-screening-ul nou-nascutilor (in 2002, 12.8% din persoanele nou diagnosticate au fost identificate prin intermediul screening-ului neonatal);
-testarea sugarilor simptomatici (cu ileus meconial), care sunt prea mici pentru a produce un volum adecvat de sudoare;
-testarea unei persoane simptomatice care are rude cu mutatii CFTR identificate6.
Deoarece mucoviscidoza se transmite autozomal recesiv, in momentul conceptiei fiecare frate al unui individ afectat are 25% sanse sa fie purtator si sa prezinte afectiunea, 50% sanse sa fie purtator asimptomatic si 25% sanse sa fie nepurtator si sa nu fie afectat.
Testarea prenatala se efectueza pe celule fetale obtinute prin biopsia vilozitatilor coriale prelevate la aproximativ 10-12 saptamani de gestatie sau prin amniocenteza, de obicei la circa 15-18 saptamani de viata intrauterina.
Testul sudorii trebuie efectuat postnatal la toti pacientii la care fibroza chistica a fost suspectata.
Testarea genetica are un rol important in detectarea unor mutatii cu implicatii importante in determinismul anumitor fenotipuri. Cea mai buna corelatie intre genotip si fenotip este legata de functia pancreatica. Mutatiile cele mai frecvente au fost clasificate in doua categorii: cele care determina insuficienta pancreatica si cele care se asociaza cu functie pancreatica normala (denumite „pancreatic sufficient”, PS). Persoanele fara afectare pancreatica au, de obicei, una sau doua alele mutante de tip PS, ce sunt dominante in ceea ce priveste fenotipul pancreatic.
In schimb, corelatia genotip-fenotip este, in general, slaba pentru bolile pulmonare in fibroza chistica. Afectiunile pulmonare in randul persoanelor cu genotipuri identice variaza foarte mult, o explicatie plauzibila fiind interventia factorilor de mediu.
Heterozigotii cu mutatii ΔF508/A455E au functie pulmonara mai buna fata de indivizii homozigoti pentru ΔF508.
Severitatea bolii pulmonare la persoanele cu una sau doua mutatii R117H depinde de lungimea regiunii poli T de la nivelul intronului 8, astfel daca bolnavul prezinta varianta 5T in configuratie cis dezvolta de obicei boli pulmonare, iar cei cu varianta 7T sau 9T au un fenotip extrem de variabil, care se poate extinde de la lipsa manifestarilor pulmonare pana la forme moderate de boala.
Deoarece mutatiile A455E si R117H sunt asociate cu functie pancreatica normala, boala pulmonara mai putin severa observata la aceste persoanele ar putea fi consecinta starii de nutritie mai bune6.
Un test genetic negativ pentru mutatiile tintite nu poate exclude boala. Deoarece in prezent sunt descrise peste 1000 mutatii, pe piata exista mai multe truse de diagnostic, care pot identifica cele mai frecvente mutatii pentru o anumita zona geografica sau grup populational. Pentru cazurile cu adevarat suspecte se poate apela la metode complexe de analiza genetica a ADN-ului (secventiere).
Colegiul American de Genetica Medicala recomanda screening-ul purtatorilor utilizand un panel care pune in evidenta 23 mutatii, in care sunt incluse majoritatea mutatiilor ce au o frecventa mai mare de 0.1% in populatia generala din SUA. Lista mutatiilor de screening poate fi insa completata cu alte mutatii pentru a imbunatati sensibilitatea detectiei pentru anumite grupuri etnice.
Testele genetice sunt disponibile pentru screening-ul persoanelor asimptomatice care doresc sa afle daca sunt sau nu purtatoare ale genei defective de fibroza chistica si implica, de obicei, interviuri pre-test, dar si consiliere privind posibilul impact al rezultatelor testelor pozitive sau negative. Acest tip de analize genetice permite parintilor sa afle daca au un risc crescut de a avea un copil cu fibroza chistica.
Screening-ul purtatorilor de fibroza chistica se recomanda urmatoarelor persoane:
– adultii care au in familie rude cu fibroza chistica;
– partenerii persoanelor cu fibroza chistica; daca un partener are fibroza chistica si celalalt este purtator al genei defective de fibroza chistica, atunci copilul va avea 50% sanse de a dezvolta boala;
– cuplurile care doresc sa conceapa copii.
Daca investigatiile arata ca o persoana este purtatoare a genei defective de fibroza chistica, este necesara si testarea partenerului. Pentru ca un copil sa dezvolte afectiunea, ambii parinti trebuie sa fie purtatori ai genei mutante. In cazul in care analizele partenerului sunt negative sunt sanse minime pentru copil sa dezvolte boala6.
Specimen recoltat – sange venos4.
Recipient de recoltare – vacutainer ce contine EDTA ca anticoagulant4.
Cantitate recoltata – 5 mL sange4.
Cauze de respingere a probei – folosirea heparinei ca anticoagulant; probe coagulate sau hemolizate4.
Stabilitate proba – 7 zile la 2-8ºC4.
Metoda
In laboratorul nostru este disponibila metoda de testare: secventierea tuturor exonilor CFTR.
Raportarea si interpretarea rezultatelor
Vor fi comunicate mutatiile depistate in gena CFTR si genotipul respectiv4.
Bibliografie
1. Felix A Ratjen MD PhD FRCP(C). Cystic Fibrosis: Pathogenesis and Future Treatment Strategies. In Respir Care 2009 May;54(5):595-605.
2. Girish D Sharma. Cystic Fibrosis, www.emedicine.medscape.com, Ref Type: Internet Communication.
3. Ioan Popa, Liviu Pop, Zagorca Popa, Casandra Cilt. Ghid de management in mucoviscidoza– (fibroza chistica).
4. Laborator Synevo. Referintele specifice tehnologiei de lucru utilizate 2010. Ref Type: Catalog.
5. Mayo Clinic/Mayo Medical Laboratories. Test Catalog. Cystic Fibrosis Mutation Analysis, 70-Mutation Panel. www.mayomedicallaboratories.com. Ref Type: Internet Communication
6. Samuel M Moskowitz, James F Chmiel, Darci L Sternen, Edith Cheng, and Garry R Cutting.CFTR- Related Disorders. Gene Reviews, 2008. www.ncbi.nlm.nih.gov. Reference Type: Internet Communication.
Produsul a fost adăugat în coș
În plus, ai la dispoziție 30 de zile pentru a veni la recoltare.