- Teste de hematologie
- Teste de biochimie
- Biochimie generală din sânge și urina
- Proteine specifice in ser si urina
- Teste biochimice din lichide de punctie
- Teste biochimice din materii fecale
- Teste biochimice pentru tulburari ereditare de metabolism
- Teste pentru nefrolitiaza
- Vitamine, oligoelemente, stres oxidativ
- Acizi grași
- Transferina carbohidrat deficitara (CDT) marker pentru alcoolism
- Markeri non-invazivi pentru afecţiunile hepatice
- Analiza chimică calculi
- Markeri endocrini
- Markeri tumorali
- Markeri virali
- Markeri cardiaci
- Markeri anemie
- Markeri ososi
- Markeri boli autoimune
- Anticorpi antispermatozoizi
- Autoanticorpi in afectiuni endocrine, cardiace, renale
- Autoanticorpi in afectiuni neurologice
- Autoanticorpi in afectiunile dermatologice
- Autoanticorpi in anemia pernicioasa
- Autoanticorpi in diabetul zaharat
- Markeri pentru afectiuni hepatice si gastrointestinale autoimune
- Markeri pentru afectiuni reumatismale si vasculite
- Markeri pentru monitorizarea evolutiei si tratamentului
- Markeri pentru sindromul antifosfolipidic
- Serologie boli infectioase
- Teste specializate de alergologie si imunologie
- Teste de biologie moleculara
- Teste de citogenetica
- Teste de microbiologie
- Toxicologie
- Citologie cervico-vaginala
- Histopatologie
- Consult genetic
- Genetica medicala

Sindrom adrenogenital (deficit 3-beta-HS-dehidrogenaza)-HSD3B2
Preț: 1755.00 lei
Informatii generale – Sindrom adrenogenital (deficit 3-beta-HS-dehidrogenaza)-HSD3B2
Hiperplazia suprarenaliana congenitala sau sindromul adrenogenital reprezinta un grup de afectiuni cu transmitere autozomal recesiva cauzate de deficitul uneia sau mai multor enzime implicate in sinteza normala a steroizilor din cele trei clase hormonale principale: mineralocorticoizi (aldosteron), glucocorticoizi (cortizol) si hormoni sexuali.
In toate formele exista scaderea productiei de cortizol ceea ce conduce la o sinteza crescuta de ACTH hipofizar; stimularea excesiva a suprarenalei determina hiperplazie asociata cu hipersecretie de steroizi si de metaboliti ai acestora proximal de defectul enzimatic.
Manifestarile clinice variaza in functie de localizarea defectului enzimatic in calea biochimica a steroizilor suprarenalieni si de gradul deficientei (vezi figura 1).
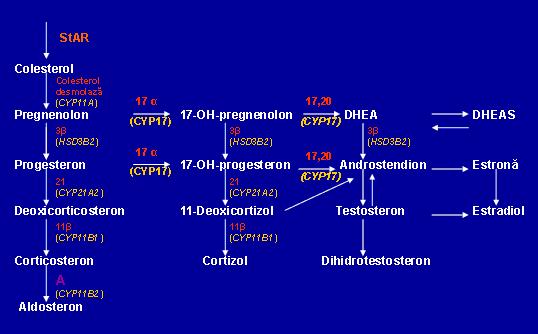
Fig. 1 Caile biochimice de sinteza a hormonilor steroizi
StAR = steroid acute regulatory protein (proteina ce transporta colesterolul la nivelul membranei mitocondriale interne)
17 α = 17α-hidroxilaza; 17,20 = 17,20-liaza; 3 β = 3β-hidroxisteroid-dehidrogenaza;
11 β = 11β-hidroxilaza; DHEA = dehidroepiandrostendion; DHEAS = DHEA sulfat
A: procesul de sinteza in 2 etape a aldosteronului:
- hidroxilarea corticosteronului pentru a forma 18-hidroxicorticosteron
- oxidarea 18-hidroxicorticosteronului pentru a forma aldosteron
(Adaptare dupa www.questdiagnostics.com)
In timp ce persoanele cu un defect enzimatic partial pot avea o simptomatologie redusa prezentand cel mai adesea un tablou clinic sugestiv pentru sindomul ovarelor polichistice, cele cu defect sever pot manifesta insuficienta suprarenaliana acuta cu pierdere de sare, ambiguitate genitala si virilizare (de la forme usoare pana la masculinizarea completa a organelor genitale externe a fetilor de sex feminin XX)9.
Tinand cont de severitatea manifestarilor clinice, sindromul adrenogenital poate fi impartit intr-o forma clasica (severa, cu debut neonatal) si o forma non-clasica (criptogena, cu debut tradiv), La randul sau forma clasica este subdivizata in varianta asociata cu pierdere de sare si in cea fara pierdere de sare (varianta virilizanta simpla).
Hormonii corticosuprarenalieni sunt derivati steroizi sintetizati din LDL-colesterol furnizat suprarenalelor si captat de receptori specifici. StAR asigura transportul acestuia prin membrana mitocondriala externa in vederea initierii cailor de sinteza a steroizilor. Enzimele care afecteaza biosinteza cortizolului sunt: P450SCC (defect la nivelul StAR), 3β-hidroxisteroid dehidrogenaza, 21-hidroxilaza, 11-hidroxilaza si 17-hidroxilaza3.
Deficitul de 21-hidroxilaza constituie cea mai frecventa cauza de sindrom adrenogenital, fiind responsabil de aproximativ 90-95% din cazuri. Enzima este localizata in reticululul endoplasmatic si catalizeaza conversia 17-OH progesteronului in 11-deoxicortizol, precum si cea a progesteronului in deoxicorticosteron (DOC). Astfel, deficitul de 21-hidroxilaza conduce la niveluri crescute de 17-OH progesteron in ser si de valori crescute de pregnanetriol si 17-OH progesteron in urina. Acesti precursori sunt suntati catre calea care conduce la o productie excesiva de androstendion si testosteron, cu virilizare consecutiva. In acelasi timp se inregistreaza scaderi variabile ale nivelurilor de DOC, 11-deoxicortizol, corticosteron, cortizol si aldosteron.
Tabloul clinic se coreleaza adesea cu severitatea disfunctiei enzimatice. Forma clasica de boala debuteaza in perioada neonatala sau in prima copilarie cu insuficienta suprarenaliana insotita de virilizare, cu sau fara pierdere de sare. Nou-nascutii de sex feminin (XX) prezinta grade variabile de masculinizare, de la marirea clitorisului pana la dezvoltarea completa a organelor genitale externe de tip masculin, ceea ce face dificila stabilirea sexului la nastere. Nou-nascutii de sex masculin au organe genitale externe normale. Aproximativ 75% din nou-nascutii afectati asociaza un deficit de aldosteron care determina pierderea de sare; simptomele de hiponatremie, hiperkaliemie, depletie a volumului sangvin si hipotensiune se manifesta in cursul primelor 2 saptamani de viata.
Forma non-clasica se manifesta mai tirziu in copilarie prin adrenarha precoce sau in perioada de adult tanar cu hirsutism, amenoree si infertilitate. La femei tabloul clinic este similar celui asociat cu sindromul ovarelor polichistice. Barbatii pot dezvolta pubertate pecoce, resturi de tesut suprarenalian in testicule si infertilitate.
Forma clasica a sindromului adrenogenital are o incidenta de 1 la 5000-15000 nasteri de feti vii, in timp ce forma non-clasica de 1 la 10003;8;9.
Exista o relatie stransa intre corticosuprarenala si medulosuprarenala. In deficitul clasic de 21-hidroxilaza a fost descrisa displazia medulosuprarenalei si scaderea productiei de catecolamine. S-a sugerat ca gradul afectarii suprarenalei ar putea constitui un marker pentru severitatea sindromului adrenogenital3.
Diagnosticul de sindrom adrenogenital prin deficit de 21-hidroxilaza este stabilit prin determinarea cantitativa a hormonilor steroizi si a precursorilor acestora. Programele de screening neonatal instituite in multe tari, care constau in masurarea nivelului de 17-OH progesteron in spoturi de sange recoltate pe hartie de filtru au scopul de a identifica nou-nascutii cu forma clasica de deficit de 21-hidroxilaza deoarece instituirea precoce a tratamentului minerolocorticoid/glucocorticoid poate preveni aparitia crizelor severe de pierdere de sare9;10.
Gena functionala pentru 21-hidroxilaza suprarenaliana – CYP21A2 – este localizata pe bratul scurt al cromozomului 6 in cluster-ul de gene HLA, la o distanta de 30 kb de o pseudogena CYP21A2P care este inactiva ca urmare a unor mutatii multiple. CYP21A2 si CYP21A2P prezinta un grad inalt de similitudine a secventelor nucleotice (98% identitate intre exoni si 96% intre introni) si ambele contin 10 exoni. Au fost descrise cinci alele normale ale genei active. Cele doua gene sunt situate intr-o regiune in care exista si alte gene duplicate, dispuse in tandem, ceea ce faciliteaza aparitia evenimetelor de recombinare intre secventele repetate. Aceste recombinari reprezinta o cauza majora de mutatii CYP21A2 ce determina deficitul de 21-hidroxilaza. Recombinarea ce rezulta ca urmare a crossing-over-ului inegal din cursul meiozei intre omologii CYP21A2 functionali poate genera o deletie sau o duplicatie mare a genei. Pe de alta parte, gradul mare de similitudine a secventelor nucleotidice intre CYP21A2 si CYP21A2P faciliteaza conversia genica, fenomen prin care un segment al genei functionale CYP21A2 este inlocuit de un segment copiat din pseudogena CYP21A2P care contine mutatii; astfel, sunt generate variante patologice care inactiveaza expresia normala a genei CYP21A2 si/sau translatia proteinei normale.
Au fost descrise pana in prezent peste 100 mutatii CYP21A2 ce includ mutatii punctiforme, deletii sau insertii mici, precum si rearanjari complexe ale genei. Majoritatea persoanelor cu deficit de 21-hidroxilaza sunt heterozigoti compusi (prezinta 2 alele mutante diferite). Cele mai obisnuite mutatii sunt reprezentate de: c.293-13A>G; c.293-13C>G, p.Pro31Leu, p.Ile173Asn, cluster-ul de mutatii ale exonului 6 p.(Ile237Asn, Va238Glu, Met240Lys), p.Val282Leu, p.Leu308PhefsX6, p.Gln319X, p.Arg357Trp, p.Pro454Ser, p.Gly111ValfsX21. Acestea impreuna cu deletia de 30 kb ce afecteaza capatul 3’ al pseudogenei CYP21A2P, gena adiacenta a complementului C4B si capatul 5’ al genei CYP21A2 constituie ~90% dintre mutatiile cauzatoare de boala.
La marea majoritate a pacientilor cu sindrom adrenogenital prin deficit de 21-hidroxilaza genotipul CYP21A2 poate fi utilizat in scop predictiv pentru evaluarea severitatii bolii. Astfel, fenotipurile asociate cu pierdere de sare, virilizare simpla, sau forma non-clasica pot fi „prognosticate” la persoanele care efectueaza teste de genetica moleculara. In general fenotipul se coreleaza cu gradul cel mai mare de activitate enzimatica reziduala de la o alela mutanta (fenotipul exprimat reflecta mutatia cu efectul fenotipic mai putin sever).
Uneori, din motive necunoscute, genotipul obtinut nu se coreleaza intotdeauna cu acelasi fenotip, fie in cazul grupurilor cu mutatii identice, fie in cadrul aceleiasi familii.
Pe baza activitatii enzimatice reziduale alelele mutante sunt clasificate in severe si usoare (vezi tabelul 2).
Genotipul asociat cu forma clasica de boala se caracterizeaza prin prezenta unei mutatii severe pe ambele alele CYP21A2. Deletiile homozigote ale genei constituie cele mai severe anomalii, fiind intotdeauna asociate cu fenotipul insotit de pierdere de sare. Conversiile genice mari pot inlocui un segment mare al genei functionale CYP21A2 cu un segment al pseudogenei CYP21A1P ce este nefunctional ca urmare a unor mutatii multiple. Acestea se manifesta de asemenea printr-un fenotip sever.
Una din cele mai frecvente mutatii (intalnita in 20-30% din cazuri) este substitutia A>G sau C>G la nivelul intronului 2 (c.293-13A>G; c.293-13C>G) care are drept rezultat un splicing aberant al ARNm si o modificare a cadrului de citire; va fi generata in final o proteina trunchiata, nefunctionala. Desi majoritatea persoanelor care sunt homozigote pentru aceasta mutatie prezinta varianta de boala cu pierdere de sare, severitatea tulburarii hidroelectrolitice este variabila. Astfel, mutatia intronului 2 poate fi intalnita si in formele virilizante simple. Aceasta lipsa de concordanta genotip-fenotip poate fi explicata prin splicing-ul alternativ crescut care apare atunci cand cel normal este suprimat ca urmare a unei mutatii a situsului de splicing, ceea ce permite o oarecare productie a proteinei cu activitate variabila. Aproximativ 20% din alelele mutante sunt rezultatul recombinarii meiotice intre secvente repetate care vor da nastere deletiei de 30 kb descrise mai sus, ce va conduce in final la o pseudogena himerica nefunctionala. In urma conversiilor genice mici pot sa mai apara urmatoarele anomalii asociate cu fenotip sever:
-o deletie de 8 baze in exonul 3 (G110del8): este afectat cadrul de citire, conducand la aparitia unui codon stop prematur (130) si, in final, la o proteina scurtata;
-mutatiile punctiforme missens de la nivelul exonului 6 (I236N, V237E, M239K);
-insertia unei baze in exonul 7 (F306+T);
-o mutatie nonsens in codonul 318 (Q318X);
-o mutatie missens in exonul 8 (R356W).
Mutatia I172N este intalnita frecvent la pacientii care prezinta forma de boala cu virilizare simpla; nu apar tulburari hidrolectrolitice deoarece exista o activitate enzimatica reziduala care asigura o productie minima de aldosteron.
Genotipul asociat cu forma non-clasica de boala contine doua mutatii usoare CYP21A2 sau o mutatie usoara si una severa. Aproximativ 2/3 din pacienti sunt heterozigoti compusi. Mutatiile missens P30L si V281L sunt caracteristice pentru pacientii care prezinta forma non-clasica, insa exista si cazuri rare (<3%) in care una dintre aceste mutatii in asociere cu o mutatie severa pot da nastere unui fenotip clasic1;10.
|
Activitate enzimatica |
Fenotip |
Mutatie CYP21A2 |
|
0% |
Sever (clasic)
|
Deletia intregii gene (mutatie nula) Conversie genica mare p.Gly111ValfsX21 (G110del8) p.[Ile237Asn; Val238Glu; Met240Lys] (I236N, V237E, M239K) p.Leu308PhefsX6 (F306+T) p.Gln319X (Q318X) p.Arg357Trp (R356W) |
|
Activitate reziduala minima (<1%) |
c.293-13A>G sau c.293C>G (In2G) |
|
|
2%-11% |
p.Ile173Asn (I172N) |
|
|
~ 20%-50% |
Usor (non-clasic) |
p.Pro31Leu (P30L) p.Val282Leu (V281L) p.Pro454Ser (P453S) |
Tabel 2. Clasificarea principalelor mutatii CYP21A2 in functie de activitatea enzimatica reziduala
Prin secventierea intregii gene CYP21A2 pot fi depistate mutatii rare: de exemplu mutatiile V304M si G375S au fost depistate la femei cu hiperandrogenism care nu prezentau nici unul dintre genotipurile asociate in mod obisnuit cu sindrom adrenogenital prin deficit de 21-hidroxilaza7.
Deficitul de 11β-hidroxilaza este al doilea defect intalnit in biosinteza de hormoni steroizi fiind responsabil de 5-8% din cazurile de sindrom adrenogenital (incidenta de 1:100 000 nou- nascuti). In cazul acestui defect enzimatic este blocata transformarea 11-deoxicortizolului in cortizol si cea a DOC in corticosteron si, consecutiv, in aldosteron. Spre deosebire de deficitul de 21-hidroxilaza, aceasta forma nu asociaza pierdere de sare insa virilizarea fetilor de sex feminin (XX) poate fi la fel de severa. Activitatea reninei plasmatice este adesea suprimata de nivelurile crecute de DOC care determina retentie hidrosalina. Din acest motiv, aproximativ doua treimi din pacienti manifesta precoce hipertensiune arteriala.
Nivelurile crescute ale tensiunii arteriale impreuna cu cele ale DOC si 11-deoxicortizolului faciliteaza diferentierea clinica intre deficitul de 11-hidroxilaza si cel de 21-hidroxilaza. Tratamentul substitutiv cu glucocorticoizi normalizeaza nivelurile DOC si activitatea reninei plasmatice (hiperaldosteronism remediabil cu glucocorticoizi).
Exista si o forma de boala cu debut tardiv asemanatoare clinic cu forma non-clasica de 21-hidroxilaza.
Gena care codifica 11-hidroxilaza – CYP11B1 – este localizata pe cromozomul 8q21, la 40 kb distanta de o gena inalt omoloaga – CYP11B2 – corespunzatoare aldosteron sintazei. Desi a fost descris un cluster de mutatii la nivelul exonilor 2, 6, 7 si 8, studiile au aratat ca mutatiile inactivatoare CYP11B1 sunt distribuite la nivelul intregii regiuni codante, ceea ce face ca strategia de testare sa includa secventierea intregii gene. Majoritatea mutatiilor sunt missens cu afectarea cadrului de citire si aparitia prematura a codonului stop. In plus au fost descrise si gene himerice, rezultate ca urmare a crossing-over-ului inegal intre CYP11B1 si CYP11B2, care suprima complet functia 11-hidroxilazei2;3;9;11.
La evreii de origine marocana, unde prevalenta acestui defect este relativ crescuta (1 la 5000-7000 nasteri de feti vii), este prezenta aproape intotdeauna aceeasi mutatie R448H in exonul 8: substitutia argininei cu histidina la nivelul codonului 448 care afecteaza activitatea enzimatica13.
Deficitul de 3β-hidroxisteroid dehidrogenaza constituie o forma rara de sindrom adrenogenital (<1% din cazuri) caracterizata prin niveluri crescute de pregnenolon, 17-OH pregnenolon, DHEA si prin niveluri scazute ale tuturor celorlalti steroizi suprarenalieni.
La om sunt descrise doua izoenzime ale 3b-hidroxisteroid dehidrogenazei, care difera doar prin 23 aminoacizi, codificate de genele HSD3B1 si, respectiv, HSD3B2 ce se exprima in tesuturi diferite. Astfel, HS23B2 se exprima la nivelul suprarenalelor si gonadelor, in timp ce HSD3B1 se exprima in placenta, la nivel cutanat, precum si in alte tesuturi periferice si, de obicei, ramane intacta in sindromul adrenogenital.
Pacientii cu forma clasica de boala vor prezenta manifestari ale deficitului de glucocorticoizi, asociate sau nu cu pierdere de sare. Baietii prezinta o masculinizare incompleta; se inregistreaza variatii fenotipice, de la hipospadias pana la pseudohermafrodism masculin. Fetitele pot fi normale sau pot prezenta virilizare usoara. A fost descrisa si o forma de boala cu debut tardiv asociata cu manifestari similare sindromului ovarelor polichistice (hirsutism, oligomenoree si infertilitate)3;4;9.
Gena HSD3B2 are o lungime de 8 kb si este localizata pe cromozomul 1 in regiunea p11-13. Au fost descrise cel putin 31 mutatii diferite in deficitul de 3b-hidroxisteroid dehidrogenaza; forma clasica de boala cu pierdere de sare in care activitatea enzimatica este suprimata complet este asociata cu mutatii missens neconservative ale cadrului de citire si ale regiunii de splicing. In formele non-clasice cu debut tardiv se inregistreaza un deficit enzimatic partial indus de anumite mutatii missens HSD3B24.
Cateva exemple de mutatii HSD3B2:
-mutatiile A10E, G15D, W171X, L205P, P222Q, P222T, R249X, 818delAA (status homozigot) au fost descrise la pacienti cu pierdere de sare;
-mutatia A82T a fost asociata cu fenotipul fara pierdere de sare; apare cu frecventa mai mare in populatia feminina;
-mutatiile T259M (status homozigot) si G129R/P222Q (status heterozigot compus) au fost descrise la femei cu virilizare5;12.
Recomandari pentru testarea genetica
-confirmarea diagnosticului la persoanele cu suspiciune clinica:
• nou-nascuti cu test screening pozitiv;
• nou-nascuti cu ambiguitate genitala;
• nou-nascuti sau copii cu insuficienta suprarenaliana si/anomalii inexplicabile ale sodiului si potasiului;
• copii cu pubertate precoce sau, dimpotriva, intarziata;
• copii cu hipertensiune arteriala fara alte cauze;
• femei cu sindrom al ovarelor polichistice, hirsutism si/sau semne de deficit estrogenic;
• persoane cu suspiciune de hiperandrogenism;
-stabilirea prognosticului persoanelor afectate (corelatii genotip-fenotip);
-depistarea purtatorilor in familiile cu istoric pozitiv pentru sindromul adrenogenital;
-diagnostic prenatal in cazul sarcinilor cu risc crescut, dupa identificarea in familie a mutatiilor cauzatoare de boala2;4;9;10.
Specimen recoltat – sange venos6.
Recipient de recoltare – vacutainer ce contine EDTA ca anticoagulant6.
Cantitate recoltata – 5 mL sange6.
Cauze de respingere a probei – folosirea heparinei ca anticoagulant; probe coagulate sau hemolizate4.
Stabilitate proba – 7 zile la 2-8ºC6.
Metode
In laboratorul nostru sunt disponibile urmatoarele tipuri de teste6:
- Testarea genetica pentru deficitul de 21-hidroxilaza: secventiere completa CYP21A2 + analiza deletiilor-duplicatiilor MLPA.
- Testarea genetica pentru deficitul de 11-hidroxilaza: secventiere completa CYP11B1.
- Testarea genetica pentru 3b-hidroxisteroid dehidrogenaza: secventiere completa HSD3B2.
Interpretarea rezultatelor
Testarea genetica pentru deficitul de 21-hidroxilaza
In absenta oricarei rearanjari, deletii sau mutatii heterozigote cunoscute ca fiind asociate cu boala, diagnosticul de deficit de 21-hidroxilaza este putin probabil.
Depistarea statusului homozigot sau heterozigot compus pentru rearanjari, deletii sau mutatii cunoscute ca fiind cauzatoare de boala confirma diagnosticul de deficit clasic de 21-hidroxilaza.
Depistarea pe o alela CYP21A2 a uneia din cele 3 mutatii cunoscute ca fiind asociata cu forma non-clasica a bolii si pe cealalta alela a unei rearanjari, deletii sau mutatii cunoscute ca fiind cauzatoare de boala confirma diagnosticul de forma non-clasica de deficit de 21-hidroxilaza.
Depistarea unei singure rearanjari, deletii sau mutatii heterozigote cunoscute ca fiind asociata cu boala denota starea de purtator8.
Majoritatea parintilor copiilor afectati de deficit de 21-hidroxilaza sunt heterozigoti pentru o alela mutanta. Heterozigotii sunt clinic asimptomatici insa pot prezenta valori usor crescute de 17-OH progesteron la stimularea cu ACTH. Este important sa fie testati ambii parinti deoarece in unele cazuri un parinte poate fi depistat ca avand forma non-clasica de boala.
In situatia in care sunt detectate mutatii multiple este posibil ca acestea sa fie prezente in configuratia trans (pe cromozomi separati, mostenite de la ambii parinti) sau in configuratia cis (situate pe acelasi cromozom ca rezultat al conversiei genice, considerate ca fiind o singura alela mutanta). Pentru evitarea erorilor de diagnostic investigarea ambilor parinti este obligatorie.
Daca pe acelasi cromozom se depisteaza prin MLPA alaturi de gena CYP21A2 functionala si o duplicatie a genei cu o mutatie pacientul nu va fi considerat purtator.
Aproximativ 1% dintre mutatii sunt de novo astfel ca 1% dintre persoanele afectate au doar un singur parinte heterozigot pentru o alela mutanta.
Daca ambii parintii sunt heterozigoti, 25% dintre copii prezinta riscul de a mosteni ambele alele si de a dezvolta boala, 50% au riscul de a deveni purtatori ai unei alele mutante si 25% prezinta ambele alele normale.
Diagnosticul prenatal in cazul sarcinilor cu risc crescut pentru forma clasica de deficit de 21-hidroxilaza este foarte important deoarece instituirea tratamentului prenatal cu dexametazona la fetii de sex feminin afectati reduce virilizarea.
Un program de tratament prenatal trebuie sa includa urmatoarele etape:
• consilierea genetica preconceptie;
• testarea genetica preconceptie atat a copilului afectat cat si a parintilor, in vederea identificarii celor 2 mutatii CYP21A2 cauzatoare de boala;
• dupa confirmarea sarcinii si inainte de 9 saptamani se va administra zilnic mamei dexametazona in 3 doze orale pentru a suprima eventualul exces androgenic fetal;
• la 10-12 saptamani de sarcina se va efectua prelevare de vilozitati coriale (de preferat) sau la 15-18 saptamani amniocenteza in vederea diagnosticului prenatal;
• daca fatul este de sex masculin sau este vorba de un fat de sex feminin neafectat se va intrerupe tratamentul cu dexametazona;
• daca fatul este de sex feminin si se constata ca prezinta mutatiile asociate cu forma clasica de sindrom adrenogenital sau este vorba de un status genetic incert, se va continua tratamentul cu dexametazona pana la termen; tratamentul prenatal nu are nici un efect asupra necesitatii de tratament hormonal substitutiv postnatal.
In general nu se recomanda tratament prenatal la fetii cu risc de forma non-clasica de sindrom adrenogenital; exista doar citeva exceptii rare de neconcordanta genotip-fenotip10.
Testarea genetica pentru deficitul de 11-hidroxilaza
Interpretarea rezultatelor este similara cu cea pentru deficitul de 21-hidroxilaza. Daca ambii parinti sunt cunoscuti a fi purtatori de mutatii CYP11B1 asociate cu forma virilizanta de boala exista optiunea tratamentului prenatal la fetii de sex feminin afectati6.
Testarea genetica pentru 3b-hidroxisteroid dehidrogenaza
Fiind de asemenea o afectiune cu transmitere autozomal-recesiva interpretarea rezultatelor este similara cu cea pentru deficitul de 21-hidroxilaza6.
Bibliografie
1. Catherine E. Keegan, Anthony A. Killeen. An Overview of Molecular Diagnosis of Steroid 21- Hydroxylase Deficiency. In JMD 2001, Vol.3, No.2.
2. Gabriel I Uwaifo. C-11 Hydroxylase Deficiency. www. emedicine.medscape.com. Ref Type: Internet Communication.
3. Helena A. Guber, Amal F. Farag, James Lo, James Sharp. Evaluation of Endocrine Function. In Henry´s Clinical Diagnosis and Management By Laboratory Methods, Ed.Saunders, 2007, 344-348.
4. J. Paul Frindik . 3-Beta-Hydroxysteroid Dehydrogenase Deficiency. www. emedicine.medscape.com. Ref Type: Internet Communication.
5. Jacques Simard, Marie-Louise Ricketts, Sébastien Gingras, Penny Soucy, F. Alex Feltus and Michael H. Melner. Molecular Biology of the 3ß-Hydroxysteroid Dehydrogenase/Δ5-Δ4 Isomerase Gene Family. In Endocrine Reviews 26 (4): 525-582.
6. Laborator Synevo. Referinte specifice tehnologiei de lucru utilizate 2017. Ref Type: Catalog.
7. Lajic S, Clauin S,Robins T, Vexiau P, Blanche H, Bellanne-Chantelot C, Wedell A. Novel mutations in CYP21 detected in individuals with hyperandrogenism. In The Journal of Clinical Endocronology and Metabolism, 2002, 87(6):2824-2829.
8. Mayo Clinic, Mayo Medical Laboratories. Reference Laboratory Services for Health Care Organizations. 21-Hydroxylase Gene, Known Mutation. www.mayomedicallaboratories.com. 2017. Ref Type: Internet Communication.
9. Quest Diagnostics. Congenital Adrenal Hyperlasia. Laboratory Support of Diagnosis and Manangement. Clinical Focus. www.questdiagnostics.com. Reference Type: Internet Communication.
10. Saroj Nimkarn, Maria I New. 21-Hydroxylase-Deficient Congenital Adrenal Hyperplasia. Gene Reviews, 2016. www.ncbi.nlm.nih.gov. ReferenceType: Internet Communication.
11. Silvia Parajes, Lourdes Loidi, Nicole Reisch, Vivek Dhir, Ian T. Rose, Rainer Hampel, Marcus Quinkler, Gerard S. Conway, Lidia Castro-Feijóo, David Araujo-Vilar, Manuel Pombo, Fernando Dominguez, Emma L. Williams, Trevor R. Cole, Jeremy M. Kirk, Elke Kaminsky, Gill Rumsby, Wiebke Arlt, Nils Krone. Functional Consequences of Seven Novel Mutations in the CYP11B1 Gene: Four Mutations Associated with Nonclassic and Three Mutations Causing Classic 11β-Hydroxylase Deficiency. In J Clin Endocrinol Metab. 2010 February; 95(2): 779-788.
12. Suemi M, Russell AJ, Paula F, Dick-de-Paula I, Marcondes JA, Mendonca BB. Genotyping of the type II 3b hydroxysteroid dehydrogenase gene (HSD3B2) in women with hirsutism and elevated ACTH-stimulated D5-steroids. In Fertiltiy and Sterility, 2000, 74 (3): 553-557.
13. White PC, Dupont J, New MI, Leibermann E, Hochberg Z, Rosler A. A mutation in CYP11B1 (Arg448-His) associated with steroid 11b-hydroxylase deficiency in Jews of Moroccan origin. In J Clin Invest. 1991, 87: 1664-1667.
Produsul a fost adăugat în coș
În plus, ai la dispoziție 30 de zile pentru a veni la recoltare.