Diagnosticul sindroamelor de aritmii ereditare II

Indicațiile testării genetice la pacienții cu aritmii cardiace ereditare
În 2011, Heart Rhythm Society (HRS)/ European Heart Rhythm Association (EHRA ) recomanda:
• Testarea genetică la pacienții cu o suspiciune clincă puternică pentru una dintre aceste afecțiuni;
• Testare genetică în cascadă pentru membrii familiei și rudele pacienților cu genotip pozitiv la oricare dintre aceste afecțiuni;
• Testarea genetică a pacienților cu suspiciune clinică de sindrom QT lung, prelungirea QT idiopatică, fără simptome și cu un interval QTc electrocardiografic documentat > 480 milisecunde (prepubertate) sau > 500 milisecunde (adulți) sau cu suspiciune clinică de tahicardie ventriculară polimorfă catecolaminergică;
• Testarea genetică nu este recomandată pentru pacienții cu modele electrocardiografice Brugada izolate de tip 2 sau 3, care nu sunt patognomonice pentru sindrom Brugada;
• Pentru supraviețuitorii unui stop cardiac: testarea pacienților cu suspiciune clinică pentru o cardiomiopatie specifică sau o patologie a canalelor ionice; în caz contrar, testarea nu este indicată;
• Pentru testarea post-mortem în cazurile de moarte subită cardiacă: se recomandă colectarea de probe de țesut pentru posibile teste viitoare, iar selecția unor paneluri specifice de testare ar trebui să fie ghidată de rezultatele autopsiei (dacă sunt disponibile) și de constatările testelor clinice pentru membrii supraviețuitori ai familiei.
Cuprins:
- Algoritm de diagnostic
- Diagnostic și management
- Sindrom Brugada – studiu de caz
- Tahicardie ventriculară polimorfică catecolaminergică (CPVT) – studiu de caz
- Sindromul de QT lung (LQTS) – studiu de caz
- Sindromul de QT scurt – studiu de caz
Algoritm de diagnostic
Musunuru et al. au propus strategii de testare genetică pentru pacienții cu un diagnostic confirmat sau suspiciune de afecțiune cardiacă ereditară. Rezultatul este detaliat in următoarea schemă de diagnostic:
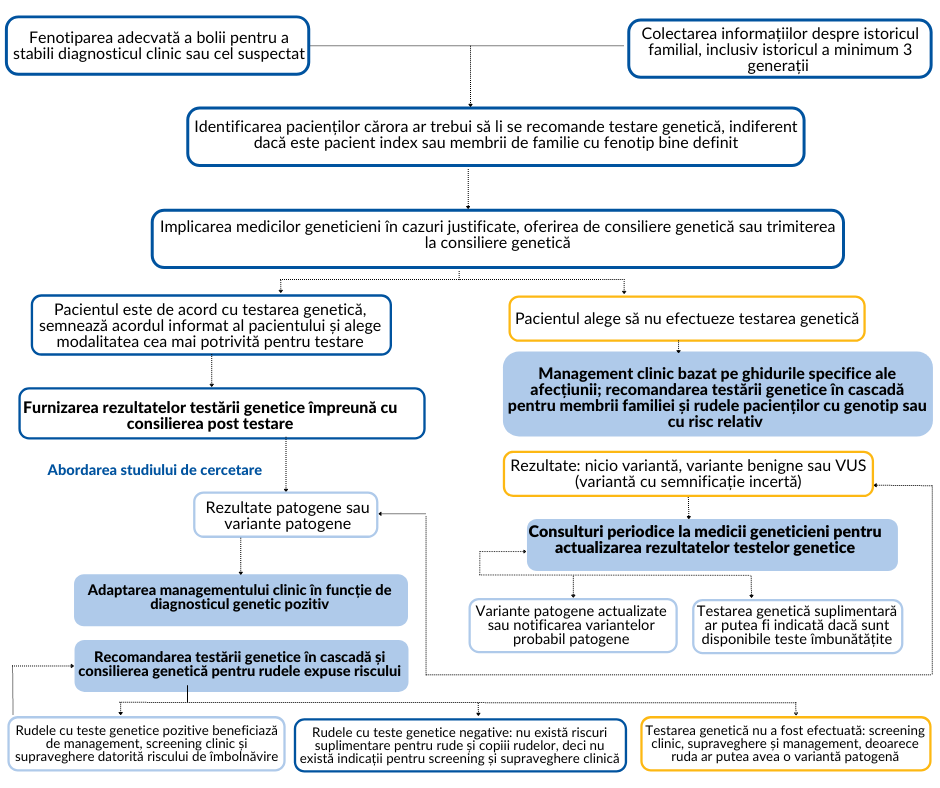
Abordarea evaluării pacienților cu diagnostic confirmat sau suspiciune de boală cardiovasculară ereditară
VUS: variantă cu semnificație incertă (din Musunuru et al. 2020)
Diagnostic și management
Diagnosticul se bazează pe prezentarea și istoricul clinic, pe caracteristicile înregistrării electrocardiografice în repaus și în timpul efortului, cât și pe analize genetice. Managementul se bazează pe terapia farmacologică, în principal blocante ale receptorilor β-adrenergici (în special, propranolol și nadolol) și blocante ale canalelor de sodiu (cum ar fi chinidina) sau intervenții chirurgicale, inclusiv ablația simpatică cardiacă stângă și implantarea unui defibrillator – cardioverter.
STUDII DE CAZ
Sindrom Brugada – studiu de caz
Un băiețel de 20 de luni a fost adus la serviciul de urgență al spitalului după ce a suferit un accident în timpul mersului cu bicicleta. Tatăl său a avut antecedente de sincopă la vârsta de 30 de ani. Testul de provocare ajmaline al tatălui a fost negativ, dar un ECG efectuat în timpul unui episod de febră a arătat un model ECG caracteristic pentru Sindrom Brugada (BrS). Sora tatălui a prezentat un model ECG sugestiv pentru BrS de tip 1 indus de ajmaline.
TESTARE, ACȚIUNI ȘI RECOMANDĂRI
I s-a efectuat ECG și pe baza rezultatelor a fost diagnosticat cu sindrom de sinus afectat (SSS) cu evadare joncțională. A fost implantat un stimulator cardiac DDD, iar în timpul implantării, atriile dilatate au fost inactive din punct de vedere electric, ceea ce a condus la diagnosticul de asistolă atrială.
În timpul recuperării, a experimentat din nou un colaps, cauzat de un tromb, care a dus la un accident vascular cerebral pe partea stângă. Ecocardiografia a arătat un contrast ecocardiografic spontan atrial stâng, fără evidențiere de trombi.
Deși în timpul monitorizării nu au fost documentate tahiaritmii ventriculare, un ECG efectuat în timpul febrei (39°C) a demascat modelul caracteristic BrS tip 1, iar pacientul a primit un defibrilator cardioverter implantabil VVI endovascular, deoarece exista un risc estimat ridicat al dezvoltării aritmiilor ventriculare.
Testarea genetică a identificat două varianteîn gena SCN5A: c.4813+3_4813+6dupGGGT, o mutație fondatoare belgiană și c.4711T>C (p.Phe1571Leu). O analiză de segregare familială a evidențiat mutația fondatoare la tată și mătușa paternă, precum și apariția de novo a variantei p.Phe1571Leu la pacientul index.
REZULTATE, ACȚIUNI POST-TESTARE GENETICĂ
BrS este rar observat în populația pediatrică și doar 4,3% dintre pacienții simptomatici prezintă un prim eveniment aritmic ventricular malign înainte de vârsta de 16 ani. Când SCN5A este afectată de două variante combinate (în stare de heterozigoție compusă), aceasta poate duce la oagravare semnificativă a simptomatologiei și/ sau la debutul mai precoce al bolii.
Acest lucru s-a observat și la acest pacient care a experimentat prima sa sincopă în timpul activității fizice în jurul vârstei de 2 ani, cauzată probabil de bradiaritmie în situația de asistola atrială.
Intervențiile cuprind, în cea mai mare parte, implantarea unui stimulator cardiac, cu adăugarea unei doze mici de aspirină sau chinidină orală și un pacient a primit beta-blocante fără implant de stimulator cardiac.
În familia prezentată, purtătorii heterozigoți ai mutației fondatoare belgiene SCN5A au prezentat un model ECG caracteristic Sindrom Brugada (BrS) tip-1 fie după provocarea cu blocant al canalelor de sodiu (mătușă), fie în timpul stărilor febrile (tată), fiind raportată sincopa la vârsta de 30 de ani (tata) sau absența simptomatologiei (mătușa).
Pe baza experimentelor funcționale, varianta p.Phe1571Leu descrisă este probabil patogenă și, în cazul prezentat, apariția ei de novo, împreună cu mutația fondatoare belgiană SCN5A, explică fenotipul sever al afecțiunilor canalelor de sodiu cardiace cu Sindrom Brugada (BrS).
Tahicardie ventriculară polimorfică catecolaminergică (CPVT) – studiu de caz
ISTORIC, PREZENTAREA CAZULUI
Un băiat caucazian de 17 ani a fost internat în unitatea de terapie intensivă (UTI) după resuscitare cu succes de către serviciile de urgență. În timp ce făcea exerciții de alergare într-un centru de fitness, s-a prăbușit brusc. Nu a avut simptome înainte de prăbușire. Cu toate acestea, în anii anteriori, a avut sincope de mai multe ori în timp ce urca scările, juca fotbal și o dată când s-a speriat. Un medic generalist a efectuat anterior un ECG de efort, care a arătat mai multe impulsuri premature sub stres submaximal.
Ca urmare, au fost prescrise beta-blocante (succinat de metoprolol 47,5 mg o dată pe zi). În afară de aceasta, pacientul nu avea antecedente patologice sau medicație administrată. Pacientul era nefumător, fără consum regulat de alcool și cu un istoric familial, social și de mediu nesemnificativ.
TESTARE, ACȚIUNI și RECOMANDĂRI
Examenele pentru diagnostic au inclus teste de sânge, angiografie coronariană, teste electrofiziologice și imagistică prin rezonanță magnetică cardiacă, dar toate rezultatele au fost normale. Deoarece istoricul medical al pacientului a inclus sincopă recurentă în timpul stresului fizic și emoțional, suspiciunea principală a fost de tahicardie ventriculară polimorfică catecolaminergică (CPVT).
Au fost efectuate teste genetice și au confirmat diagnosticul de tahicardie ventriculară polimorfică catecolaminergică (CPVT), dezvăluind o mutație punctiformă heterozigotă de novo în gena pentru RyR2, c.12520T>A (p.F4174 l, exon 90).
REZULTATE, ACTIUNI POST-TESTARE GENETICĂ
Tahicardie ventriculară polimorfică catecolaminergică (CPVT) este o boală ereditară care se prezintă cu sincopă recurentă în timpul efortului și emoțiilor acute. Acest caz de CPVT este asociat unei mutații punctiforme de novo a genei RyR2, 45% dintre mutațiile relevante ale RyR fiind localizate în această regiune.
Screening-ul genetic al ambilor părinți a arătat că niciunul dintre ei nu era purtător al mutației. Rezultatele evaluărilor suplimentare ale pacientului și membrilor familiei pentru alte mutații patogene (sindrom QT lung sau scurt, sindrom Brugada) au fost, de asemenea, negative.
Pacientul a primit un dispozitiv cardioverter-defibrilator (ICD) implantabil pentru a-l proteja de episoade recurente de fibrilație ventriculară (FV). În plus, medicația orală cu un beta-blocant a fost continuată cu doza maximă tolerabilă. La scurt timp după implantare, ICD a finalizat un episod de tahicardie ventriculară, susținut prin stimulare anti-tahicardică.
Acest episod a avut loc dimineața devreme, în timp ce pacientul dormea. În următoarele 6 luni, un singur episod de FV a apărut în timpul unei activități fizice (ciclism), de data aceasta terminându-se spontan.
Sindromul de QT lung (LQTS) – studiu de caz
ISTORIC, PREZENTAREA CAZULUI
O femeie de 30 de ani a fost inițial diagnosticată cu epilepsie, dar testul mesei înclinate (TILT) a arătat un răspuns vasodepresiv. Midodrina a fost prescrisă, dar fără a avea o ameliorare clinică. Pacienta a fost trimisă la consult cardiologic din cauza continuării sincopelor recurente; ea a raportat episoade de presincopă și sincopă, ocazional precedate de palpitații și uneori declanșate de ortostatism prelungit. Nu a existat un alt istoric medical relevant. Antecedentele familiale au inclus moartea subită a mamei sale la vârsta de 46 de ani.
TESTARE, ACȚIUNI ȘI RECOMANDĂRI
Nicio anomalie nu a fost detectată la testele de laborator și la ecocardiografia transtoracică nu au fost evidențiate modificări semnificative. Electrocardiograma (ECG) cu 12 derivații efectuată la consultația cardiologică a evidențiat ritm sinusal și prelungire semnificativă a QTc (500–565 ms).
Testarea genetică a identificat mutația c.785delG, în status heterozigot în exonul 4 al genei KCNH2, ceea ce duce la formarea unui codon STOP prematur la poziția 359 a lanțului polipeptidic (p.Gly262AlafsX98). Alte două mutații au fost găsite: c.535G>A, în status heterozigot, în exonul 3 al genei KCNQ1 (p.Gly179Ser) și c.3068G>A, de asemenea în status heterozigot, în exonul 17 al genei SCN5A (p.Arg1023His).
REZULTATE, ACȚIUNI POST-TESTARE GENETICĂ
LQTS este o afecțiune ereditară rară la pacienții fără o boală cardiacă structurală, care este asociată cu un risc ridicat de tahicardie ventriculară malignă (TV) și moarte subită cardiacă.
Pacientul a prezentat mutații în trei gene diferite. Mutația p.Gly262AlafsX98 în gena KCNH2 a fost descrisă la pacienții cu LQTS și este cunoscută ca și cauză a afecțiunii. Mutațiile p.Gly179Ser în KCNQ1 și p.Arg1023His în SCN5A au fost, de asemenea, raportate în LQTS.
Având în vedere rezultatul anterior al testului mesei inclinate (TILT) și simptomele nespecifice, s-a decis implantarea unui aparat de monitorizare a evenimentelor cardiace (Holter). Pacienta a raportat mai multe episoade de pre-sincopă, care au corespuns unor perioade de tahicardie sinusală, iar într-una dintre acestea s-a înregistrat o TV polimorfă nesusţinută.
Prin urmare, s-a decis implantarea unui cardioverter-defibrilator implantabil. La aproximativ o lună după procedură, monitorizarea de la distanță a înregistrat un șoc specific pentru TV polimorfă. Terapia ei cu beta-blocante a fost schimbată cu propanolol.
Sindromul de QT scurt – studiu de caz
ISTORIC, PREZENTAREA CAZULUI
Un băiețel de 6 luni a fost testat pentru prima dată după ce tatăl său a decedat ca urmare a unui eveniment cardiac subit (SCD / Sudden Cardiac Death), declansat de o fibrilație ventriculara (FV). ECG in 12 derivații a evidențiat un QTc < 320 ms scurtat în acel moment. Pacientul a suferit o sincopă la vârsta de 1 an.
S-a efectuat studiul electrofiziologic și a fost implantat un ICD intravenos la vârsta de 16 ani din cauza istoricului familial puternic de SCD. Cea mai recentă descărcare adecvată a ICD a fost la vârsta de 17 ani din cauza FV. La vârsta de 19 ani, a avut două terapii inadecvate din cauza supradenivelarii undei T. După ajustarea pragului de detectare și inițierea terapiei antiaritmice cu chinidină, nu au existat alte episoade de șocuri inadecvate.
Tatăl său avea 28 de ani când a murit de SCD. Nu i s-au efectuat niciodată ECG, imagistică cardiacă sau teste genetice. S-a raportat că si bunica lui paternă avea un QTc scurtat. Frații tatălui său au beneficiat toți de examinări cardiovasculare, inclusiv TTE, și niciunul dintre ei nu a avut intervale QTc scurtate sau alte identificari patologice.
TESTARE, ACȚIUNI, RECOMANDĂRI
Testarea genetică a identificat o mutație patogenă în gena KCNH2 (c.1764C>G / p.Asn588Lys). Această mutație a fost estimată a avea semnificație clinică patogenă, conform SIFT (Deleterious) sau semnificație clinică posibil patogenă conform Polyphen2 (Probably Damaging) și a fost asociată anterior cu SQTS-1. Nu au fost găsite alte mutații în genele asociate cu SQTS sau SCD.
REZULTATE, ACȚIUNI POST-TESTARE GENETICĂ
Mutația KCNH2 c.1764C>G din familia sindroamelor SQTS-1 a fost descrisă ca o mutație cu câștig de funcție a canalului hERG asociat cu SQTS-1.
Cel mai recent episod de FV la vârsta de 17 ani nu a fost legat de efort. În 2015, ICD-ul intravenos a fost înlocuit din cauza epuizării bateriei. În timpul urmăririi cazului, pacientul a suferit fractură. S-a încercat o extracție transvenoasă, dar nu a avut succes din cauza aderențelor severe în vena cavă superioară. Prin urmare, a fost implantat un ICD subcutanat. Pacientul a avut rezultate normale de TTE. Cel mai recent ECG său, la vârsta de 28 de ani a arătat un QTc de 304 ms.
Bibliografie:
- Ackerman MJ, Priori SG, Willems S, Berul C, Brugada R, Calkins H, Camm AJ, Ellinor PT, Gollob M, Hamilton R, Hershberger RE, Judge DP, Le Marec H, McKenna WJ, Schulze-Bahr E, Semsarian C, Towbin JA, Watkins H, Wilde A, Wolpert C, Zipes DP. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm. 2011 Aug;8(8):1308-39. doi: 10.1016/j.hrthm.2011.05.020. PMID: 21787999. https://www.heartrhythmjournal.com/article/S1547-5271(11)00607-2/fulltext
- Akdis D, Saguner AM, Medeiros-Domingo A, Schaller A, Balmer C, Steffel J, Brunckhorst C, Duru F. Multiple clinical profiles of families with the short QT syndrome. Europace. 2018 Jun 1;20(FI1):f113-f121. doi: 10.1093/europace/eux186. PMID: 29016797. https://academic.oup.com/europace/article/20/FI1/f113/3979526
- Bezzina CR, Barc J, Mizusawa Y, Remme CA, Gourraud JB, Simonet F, Verkerk AO, Schwartz PJ, Crotti L, Dagradi F, Guicheney P, Fressart V, Leenhardt A, Antzelevitch C, Bartkowiak S, Borggrefe M, Schimpf R, Schulze-Bahr E, Zumhagen S, Behr ER, Bastiaenen R, Tfelt-Hansen J, Olesen MS, Kääb S, Beckmann BM, Weeke P, Watanabe H, Endo N, Minamino T, Horie M, Ohno S, Hasegawa K, Makita N, Nogami A, Shimizu W, Aiba T, Froguel P, Balkau B, Lantieri O, Torchio M, Wiese C, Weber D, Wolswinkel R, Coronel R, Boukens BJ, Bézieau S, Charpentier E, Chatel S, Despres A, Gros F, Kyndt F, Lecointe S, Lindenbaum P, Portero V, Violleau J, Gessler M, Tan HL, Roden DM, Christoffels VM, Le Marec H, Wilde AA, Probst V, Schott JJ, Dina C, Redon R. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet. 2013 Sep;45(9):1044-9. doi: 10.1038/ng.2712. Epub 2013 Jul 21. Erratum in: Nat Genet. 2013 Nov;45(11):1409. Borggrefe, Martin [added]; Schimpf, Rainer [added]. PMID: 23872634; PMCID: PMC3869788. https://www.nature.com/articles/ng.2712
- Devalla HD, Gélinas R, Aburawi EH, Beqqali A, Goyette P, Freund C, Chaix MA, Tadros R, Jiang H, Le Béchec A, Monshouwer-Kloots JJ, Zwetsloot T, Kosmidis G, Latour F, Alikashani A, Hoekstra M, Schlaepfer J, Mummery CL, Stevenson B, Kutalik Z, de Vries AA, Rivard L, Wilde AA, Talajic M, Verkerk AO, Al-Gazali L, Rioux JD, Bhuiyan ZA, Passier R. TECRL, a new life-threatening inherited arrhythmia gene associated with overlapping clinical features of both LQTS and CPVT. EMBO Mol Med. 2016 Dec 1;8(12):1390-1408. doi: 10.15252/emmm.201505719. PMID: 27861123; PMCID: PMC5167130. https://www.embopress.org/doi/full/10.15252/emmm.201505719
- Fernandes M, Martins Ribeiro S, Sanfins V, Lourenço A. Long QT syndrome with mutations in three genes: A rare case. Rev Port Cardiol. 2015 May;34(5):359.e1-5. English, Portuguese. doi: 10.1016/j.repc.2014.10.004. Epub 2015 Apr 29. PMID: 25935074. https://www.revportcardiol.org/en-long-qt-syndrome-with-mutations-articulo-S217420491500077X
- Hofman N, Tan HL, Alders M, Kolder I, de Haij S, Mannens MM, Lombardi MP, Dit Deprez RH, van Langen I, Wilde AA. Yield of molecular and clinical testing for arrhythmia syndromes: report of 15 years’ experience. Circulation. 2013 Oct 1;128(14):1513-21. doi: 10.1161/CIRCULATIONAHA.112.000091. Epub 2013 Aug 20. PMID: 23963746. https://www.ahajournals.org/doi/10.1161/circulationaha.112.000091
- Koopmann TT, Alders M, Jongbloed RJ, Guerrero S, Mannens MM, Wilde AA, Bezzina CR. Long QT syndrome caused by a large duplication in the KCNH2 (HERG) gene undetectable by current polymerase chain reaction-based exon-scanning methodologies. Heart Rhythm. 2006 Jan;3(1):52-5. doi: 10.1016/j.hrthm.2005.10.014. PMID: 16399053. https://www.heartrhythmjournal.com/article/S1547-5271(05)02174-0/fulltext
- Le Scouarnec S, Karakachoff M, Gourraud JB, Lindenbaum P, Bonnaud S, Portero V, Duboscq-Bidot L, Daumy X, Simonet F, Teusan R, Baron E, Violleau J, Persyn E, Bellanger L, Barc J, Chatel S, Martins R, Mabo P, Sacher F, Haïssaguerre M, Kyndt F, Schmitt S, Bézieau S, Le Marec H, Dina C, Schott JJ, Probst V, Redon R. Testing the burden of rare variation in arrhythmia-susceptibility genes provides new insights into molecular diagnosis for Brugada syndrome. Hum Mol Genet. 2015 May 15;24(10):2757-63. doi: 10.1093/hmg/ddv036. Epub 2015 Feb 3. PMID: 25650408. https://academic.oup.com/hmg/article/24/10/2757/622868
- Makita N, Yagihara N, Crotti L, Johnson CN, Beckmann BM, Roh MS, Shigemizu D, Lichtner P, Ishikawa T, Aiba T, Homfray T, Behr ER, Klug D, Denjoy I, Mastantuono E, Theisen D, Tsunoda T, Satake W, Toda T, Nakagawa H, Tsuji Y, Tsuchiya T, Yamamoto H, Miyamoto Y, Endo N, Kimura A, Ozaki K, Motomura H, Suda K, Tanaka T, Schwartz PJ, Meitinger T, Kääb S, Guicheney P, Shimizu W, Bhuiyan ZA, Watanabe H, Chazin WJ, George AL Jr. Novel calmodulin mutations associated with congenital arrhythmia susceptibility. Circ Cardiovasc Genet. 2014 Aug;7(4):466-74. doi: 10.1161/CIRCGENETICS.113.000459. Epub 2014 Jun 10. PMID: 24917665; PMCID: PMC4140998. https://www.ahajournals.org/doi/10.1161/CIRCGENETICS.113.000459
- Mazzanti A, Kanthan A, Monteforte N, Memmi M, Bloise R, Novelli V, Miceli C, O’Rourke S, Borio G, Zienciuk-Krajka A, Curcio A, Surducan AE, Colombo M, Napolitano C, Priori SG. Novel insight into the natural history of short QT syndrome. J Am Coll Cardiol. 2014 Apr 8;63(13):1300-1308. doi: 10.1016/j.jacc.2013.09.078. Epub 2013 Nov 28. PMID: 24291113; PMCID: PMC3988978. https://www.sciencedirect.com/science/article/pii/S0735109713062827
- Mohler PJ, Splawski I, Napolitano C, Bottelli G, Sharpe L, Timothy K, Priori SG, Keating MT, Bennett V. A cardiac arrhythmia syndrome caused by loss of ankyrin-B function. Proc Natl Acad Sci U S A. 2004 Jun 15;101(24):9137-42. doi: 10.1073/pnas.0402546101. Epub 2004 Jun 3. PMID: 15178757; PMCID: PMC428486. https://www.pnas.org/content/101/24/9137.long
- Musunuru K, Hershberger RE, Day SM, Klinedinst NJ, Landstrom AP, Parikh VN, Prakash S, Semsarian C, Sturm AC; American Heart Association Council on Genomic and Precision Medicine; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular and Stroke Nursing; and Council on Clinical Cardiology. Genetic Testing for Inherited Cardiovascular Diseases: A Scientific Statement From the American Heart Association. Circ Genom Precis Med. 2020 Aug;13(4):e000067. doi: 10.1161/HCG.0000000000000067. Epub 2020 Jul 23. PMID: 32698598. https://www.ahajournals.org/doi/full/10.1161/HCG.0000000000000067
- Nijak A, Labro AJ, De Wilde H, Dewals W, Peigneur S, Tytgat J, Snyders D, Sieliwonczyk E, Simons E, Van Craenenbroeck E, Schepers D, Van Laer L, Saenen J, Loeys B, Alaerts M. Compound Heterozygous SCN5A Mutations in Severe Sodium Channelopathy With Brugada Syndrome: A Case Report. Front Cardiovasc Med. 2020 Jul 24;7:117. doi: 10.3389/fcvm.2020.00117. PMID: 32850980; PMCID: PMC7396896. https://www.frontiersin.org/articles/10.3389/fcvm.2020.00117/full
- Offerhaus JA, Bezzina CR, Wilde AAM. Epidemiology of inherited arrhythmias. Nat Rev Cardiol. 2020 Apr;17(4):205-215. doi: 10.1038/s41569-019-0266-2. Epub 2019 Oct 3. PMID: 31582838. https://www.nature.com/articles/s41569-019-0266-2
- Probst V, Wilde AA, Barc J, Sacher F, Babuty D, Mabo P, Mansourati J, Le Scouarnec S, Kyndt F, Le Caignec C, Guicheney P, Gouas L, Albuisson J, Meregalli PG, Le Marec H, Tan HL, Schott JJ. SCN5A mutations and the role of genetic background in the pathophysiology of Brugada syndrome. Circ Cardiovasc Genet. 2009 Dec;2(6):552-7. doi: 10.1161/CIRCGENETICS.109.853374. Epub 2009 Sep 29. PMID: 20031634. https://www.ahajournals.org/doi/10.1161/CIRCGENETICS.109.853374
- Schwartz PJ, Ackerman MJ, Antzelevitch C, Bezzina CR, Borggrefe M, Cuneo BF, Wilde AAM. Inherited cardiac arrhythmias. Nat Rev Dis Primers. 2020 Jul 16;6(1):58. doi: 10.1038/s41572-020-0188-7. PMID: 32678103; PMCID: PMC7935690. https://www.nature.com/articles/s41572-020-0188-7
- Seidlmayer LK, Riediger F, Pagonas N, Nordbeck P, Ritter O, Sasko B. Description of a novel RyR2 mutation in a juvenile patient with symptomatic catecholaminergic polymorphic ventricular tachycardia in sleep and during exercise: a case report. J Med Case Rep. 2018 Oct 9;12(1):298. doi: 10.1186/s13256-018-1825-6. PMID: 30296944; PMCID: PMC6176516. https://jmedicalcasereports.biomedcentral.com/articles/10.1186/s13256-018-1825-6
- Tester DJ, Arya P, Will M, Haglund CM, Farley AL, Makielski JC, Ackerman MJ. Genotypic heterogeneity and phenotypic mimicry among unrelated patients referred for catecholaminergic polymorphic ventricular tachycardia genetic testing. Heart Rhythm. 2006 Jul;3(7):800-5. doi: 10.1016/j.hrthm.2006.03.025. Epub 2006 Mar 28. PMID: 16818210. https://www.heartrhythmjournal.com/article/S1547-5271(06)01327-0/fulltext
- Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005 May;2(5):507-17. doi: 10.1016/j.hrthm.2005.01.020. PMID: 15840476. https://www.heartrhythmjournal.com/article/S1547-5271(05)00191-8/fulltext
- Tester DJ, Will ML, Haglund CM, Ackerman MJ. Effect of clinical phenotype on yield of long QT syndrome genetic testing. J Am Coll Cardiol. 2006 Feb 21;47(4):764-8. doi: 10.1016/j.jacc.2005.09.056. Epub 2006 Jan 26. PMID: 16487842. https://www.sciencedirect.com/science/article/pii/S0735109705028007
- Westenskow P, Splawski I, Timothy KW, Keating MT, Sanguinetti MC. Compound mutations: a common cause of severe long-QT syndrome. Circulation. 2004 Apr 20;109(15):1834-41. doi: 10.1161/01.CIR.0000125524.34234.13. Epub 2004 Mar 29. PMID: 15051636. https://www.ahajournals.org/doi/10.1161/01.CIR.0000125524.34234.13