- Teste de hematologie
- Teste de biochimie
- Biochimie generală din sânge și urina
- Proteine specifice in ser si urina
- Teste biochimice din lichide de punctie
- Teste biochimice din materii fecale
- Teste biochimice pentru tulburari ereditare de metabolism
- Teste pentru nefrolitiaza
- Vitamine, oligoelemente, stres oxidativ
- Acizi grași
- Transferina carbohidrat deficitara (CDT) marker pentru alcoolism
- Markeri non-invazivi pentru afecţiunile hepatice
- Analiza chimică calculi
- Markeri endocrini
- Markeri tumorali
- Markeri virali
- Markeri cardiaci
- Markeri anemie
- Markeri ososi
- Markeri boli autoimune
- Anticorpi antispermatozoizi
- Autoanticorpi in afectiuni endocrine, cardiace, renale
- Autoanticorpi in afectiuni neurologice
- Autoanticorpi in afectiunile dermatologice
- Autoanticorpi in anemia pernicioasa
- Autoanticorpi in diabetul zaharat
- Markeri pentru afectiuni hepatice si gastrointestinale autoimune
- Markeri pentru afectiuni reumatismale si vasculite
- Markeri pentru monitorizarea evolutiei si tratamentului
- Markeri pentru sindromul antifosfolipidic
- Serologie boli infectioase
- Teste specializate de alergologie si imunologie
- Teste de biologie moleculara
- Teste de citogenetica
- Teste de microbiologie
- Toxicologie
- Citologie cervico-vaginala
- Histopatologie
- Consult genetic
- Genetica medicala

Neoplazie endocrina multipla tip 2-mutatii RET (exoni 10, 11, 13, 14, 15, 16)
Preț: 2155.00 lei
Neoplazia endocrina multipla de tip 2 (MEN 2) este o afectiune ereditara rara si complexa, caracterizata prin asocierea la acelasi pacient a carcinomului tiroidian medular (CTM), feocromocitomului uni- sau bilateral si a altor hiperplazii sau neoplazii ale diferitelor tesuturi endocrine.
MEN 2 este o afectiune cu transmitere autozomal dominanta cauzata de mutatii in gena RET, localizata pe cromozomul 10q11.2.
Au fost descrise doua forme diferite de MEN 2: sporadica si familiala. Forma sporadica asociaza doua dintre principalele tumori endocrine MEN tip 2. Forma familiala, mai frecventa, reprezinta subtipul in care exista cel putin o ruda de gradul 1 cu una dintre tumorile endocrine caracteristice sindromului.
MEN 2 include trei subtipuri: MEN 2A, MEN 2B si carcinomul medular tiroidian familial (CMTF).
Sindromul MEN 2 a fost raportat la aproximativ 500–1000 familii in intreaga lume, iar prevalenta este estimata la 1 la 30000 de indivizi3.
Toate cele 3 subtipuri MEN1 prezinta un risc crescut de dezvoltare a carcinomului medular tiroidian; subtipurile 2A si 2B asociaza un risc crescut de feocromocitom; in plus, subtipul 2A este asociat cu un risc crescut de adenom sau hiperplazie paratiroidiana.
MEN 2B include si anomalii constitutionale, cum ar fi habitusul marfanoid, precum si neuroame mucoase labiale sau linguale si ganglioneuromatoza intestinala, in timp ce afectiunile paratiroidiene sunt absente4.
Debutul carcinomului tiroidian medular este tipic in prima copilarie in cadrul MEN2B, la inceputul perioadei de adult in MEN2A si la varsta mijlocie in CMTF. Astfel, forma MEN 2B este considerata mai agresiva decat MEN 2A sau CMTF deoarece boala apare mult mai devreme (o medie de 10 ani mai devreme) decat MEN 2A6.
● MEN 2A (sindromul Sipple) este forma cea mai frecventa a sindromului MEN 2 (80% din cazuri). Se caracterizeaza prin prezenta carcinomului medular tiroidian (CMT), feocromocitomului uni- sau bilateral (in peste 50% din cazuri) si hiperparatiroidismului (in 15-30% din cazuri), dar fara tumori pancreatice endocrine .
Hiperplazia celulelor C apare precoce in timpul vietii si poate fi considerata ca o leziune precursoare pentru CMT. CMT este, in general, prima manifestare a MEN 2A. In familiile cu MEN 2A, manifestarile biochimice ale CMT apar la varste intre 5-25 ani.
Feocromocitomul este o tumora secretanta de catecolamine care se dezvolta pe o hiperplazie a medulosuprarenalei, secundar unei mutatii RET a liniei germinale, dar care devine manifesta (teste biochimice si/sau metode imagistice) la doar 50% dintre pacienti. In 70% din cazuri este bilateral. Varful de incidenta maxima este in jurul varstei de 40 ani, dar pot fi interesati si copii sub 10 ani.
Mai putin de 25% din pacienti prezinta un hiperparatiroidism franc. Unele variante de MEN 2A pot fi asociate cu sindroame paraneoplazice (amiloidoza licheniforma cutanata sau producerea excesiva de ACTH).
● MEN 2B (sindromul Shimcke) se intalneste in aproximativ 5% din cazurile cu MEN 2 si include carcinomul medular tiroidian (bilateral), ganglioneuromatoza, neurofibroame, anomalii scheletale (pectus excavatum, sindrom Marfan etc.). Acest subtip MEN 2B este caracterizat prin aparitia precoce (de obicei cu 10 ani mai devreme ca MEN 2A) a unor forme mai agresive de CMT si feocromocitom (40-50 % din cazuri), neuroame multiple si/sau ganglioneuromatoza digestiva (40% dintre cazuri), dar fara interesare paratiroidiana.
● Carcinomul medular tiroidian familial (CMTF) este un subtip al MEN 2 in care persoanele afectate dezvolta numai carcinom tiroidian medular, fara alte manifestari de MEN 2. Aceasta forma se refera numai la aparitia CMT la cel putin 4 membri ai aceleiasi familii. Prognosticul este relativ bun in majoritatea cazurilor.
Cele mai multe cazuri de carcinom tiroidian medular (CMT) si/sau feocromocitom sunt sporadice. Numai 10% din cazuri sunt ereditare si se incadreaza in MEN 2.
Carcinomul medular tiroidian dezvoltat din celulele parafoliculare C producatoare de calcitonina este obligatoriu in toate cazurile de MEN 2, reprezentand de regula si prima manifestare clinica. CMT apare de obicei intr-o ordine descrescatoare a severitatii in MEN 2B, MEN 2A si CMTF. CMT se dezvolta initial pe o hiperplazie multifocala a celulelor C, a carei progresie catre CMT este extrem de variabila si poate dura mai multi ani. CMT prezinta o tendinta naturala catre metastazarea locala (limfonoduli cervicali, mediastinali) si la distanta (ficat, os, plamani).
CMT se coreleaza in general cu niveluri crescute serice ale calcitoninei (bazal sau stimulat cu pentagastrina si/sau calciu), considerata marker tumoral specific pentru CMT (normal <10 pg/mL).
Metodele imagistice (ecografia, CT, IRM) pot fi utilizate pentru a determina extensia tumorala si existenta unor posibile metastaze.
Chirurgia este tratamentul de electie al CMT, atat pentru pacientii MEN 2A, cat si pentru cei MEN 2B. Tratamentul chirurgical consta in tiroidectomie totala si limfadenectomie care, ideal, trebuie realizat inainte de varsta posibilei transformari maligne. Biopsia cu ac fin si evaluarea nivelului calcitoninei serice (bazal si la 2, respectiv 5 minute dupa stimularea cu calciu) sunt importante pentru diagnosticul preoperator al CMT.
Feocromocitomul apare la aproximativ 50% dintre pacientii MEN 2A si MEN 2B, este aproape totdeauna benign, are tendinta de a fi bilateral in 50-80% dintre cazuri. In general, feocromocitomul este prima manifestare a bolii in 25% dintre cazuri, dupa CTM care este raportat a fi sindromul de debut la 40% din bolnavi; in timp ce in 35% dintre cazuri CTM si feocromocitomul sunt diagnosticate in acelasi timp. Feocromocitomul poate determina hipertensiune arteriala, cefalee episodica, palpitatii, iritabilitate, transpiratii, paloare din cauza sintezei excesive de epinefrina, norepinefrina si dopamina de catre celulele cromafine ale glandei suprarenale.
Testele pentru metanefrinele plasmatice si urinare sunt cele mai fidele pentru screening-ul feocromocitomului; dintre investigatiile imagistice sunt indicate CT, IRM, tomografie cu metaiodobenzilguanidin (MIBG), OctreoScan-ul, tomografia prin emisie de pozitroni (positive emission tomography – PET).
Inainte de operatie, prezenta unui feocromocitom functional trebuie exclusa prin analize biochimice adecvate la toti pacientii MEN 2A si MEN 2B. Daca se descopera un feocromocitom, suprarenalectomia trebuie realizata inainte de tiroidectomie sau de alta interventie chirurgicala, pentru a evita criza catecolaminica intraoperatorie. Tratamentul feocromocitomului consta in excizia chirurgicala laparoscopica. Tratamentul pe termen lung cu alfa si beta blocante trebuie folosit doar la pacientii cu tumori nerezecabile.
Hiperparatiroidismul primar (HPTP) apare in 20-30% dintre pacientii MEN 2A si este asimptomatic in majoritatea cazurilor. Diagnosticul este pus pe baza testelor biochimice care arata un nivel ridicat al PTH si al calciului seric. Tratamentul HPTP in MEN 2 este chirurgical: paratiroidectomie subtotala sau totala cu autotransplantarea in muschiul sternocleidomastoidian sau in musculatura antebratului3.
Gena responsabila de MEN 2 este varianta patologica a protooncogenei RET. Protooncogena RET are o lungime de 80 kb si codifica un receptor protein kinazic cu rol important in proliferarea, migrarea, diferentierea si dezvoltarea celulara in timpul embriogenezei.
Proteina RET este constituita din 3 domenii: extracelular, transmembranar si intracelular. Domeniul extracelular contine 4 domenii de adeziune celulara dependente de Ca2+ „cadherin-like” (care induc si stabilizeaza modificarile conformationale necesare pentru interactiunea cu liganzi si coreceptori) si o regiune juxtamembranara bogata in cisteina (responsabila pentru structura tertiara si formarea de dimeri). Domeniul extracelular contine de asemenea mai multe situsuri de glicozilare; proteina glicozilata, forma matura RET, cu o masa moleculara de 170 kDa este localizata in membrana celulara; forma imatura, neglicozilata, de 150 kDA, este prezenta numai in reticulul endoplasmic si citoplasma. Domeniul intracelular include doua subdomenii de tirozin-kinaza (TK1 si TK2) care sunt implicate in activarea a numeroase cai de transductie a semnalelor intracelulare. In functie de splicing-ul alternativ al regiunii 3’ se genereaza 3 izoforme ale proteinei care contin 9 (RET9), 43 (RET43) si, respective, 51 (RET51) aminoacizi la capatul carboxi-terminal; RET9 si RET 51 sunt principalele izoforme (vezi fig.).
In mod normal RET poate fi activata de catre un complex de coreceptori si liganzi ce apartin a doua grupe de proteine: liganzi din familia factorului neurotrofic de origine gliala (GDNF) si α-receptori pentru familia GDNF. La cuplarea ligandului receptorii RET normali dimerizeaza, iar aceasta dimerizare activeaza functia kinazica a RET si transductia de semnale activatoare. Mutatiile activatoare la nivelul genei RET induc activarea oncogenica a domeniului tirozin-kinazic cu rol semnificativ in dezvoltarea tumorilor neuroendocrine.
RET are expresie specifica in celulele derivate din creasta neurala, celulele C din glanda tiroida sau celulele cromafine din suprarenala. In MEN2 au fost depistate mutatii punctiforme in gena RET la nivelul liniei germinale; identificarea prin testare genetica a persoanelor cu risc crescut a permis instituirea unor masuri eficace de profilaxie.
Aproximativ 98% dintre pacientii MEN 2 prezinta mutatii ale c-RET afectand mai multi dintre cei 21 exoni ai genei. Mutatiile MEN 2 sunt localizate in exonii 10, 11, 13, 14, 15, 16, si 8. Mutatiile c-RET de la nivelul domeniului bogat in cisteina si de la nivelul domeniilor tirozin-kinazice determina activarea constitutiva a tirozin-kinazei receptorului mutant. Mecanismele oncogenetice ale diferitelor mutatii RET par a fi dependente de pozitia de substitutie a aminoacidului. Astfel, mutatiile de la nivelul domeniului extracelular bogat in cisteina, intalnite in general la pacientii cu MEN 2A, transforma un reziduu cisteinic intr-unul non-cisteinic. In mod normal aceste reziduuri de cisteina sunt implicate in formarea de legaturi disulfidice intramoleculare in proteina RET salbatica; ca urmare a mutatiilor un reziduu de cisteina necuplata dintr-un monomer de RET formeaza o legatura disulfidica intermoleculara cu un alt monomer mutant. Cele doua molecule RET mutante sunt activate si dimerizate constitutiv in trans, indiferent de prezenta ligandului. Mutatiile cisteinei din pozitia 634 au demonstrat o capacitate mai mare de transformare maligna comparativ cu mutatiile altor reyiduuri extracelulare de cisteina.
Pe de alta parte marea majoritate a mutatiilor la pacientii cu MEN 2B si CMTF afecteaza unul dintre cele doua domenii intracelulare cu rol de tirozinkinaza si altereaza specificitatea de substrat a acestora prin modificari ale domeniului de legare. Rezulta o fosforilare aberanta a substraturilor preferate de tirozin kinazele citoplasmatice, ca de exemplu c-Src si c-abl, in detrimentul substratului folosit in mod normal de receptorul tirozin kinazic.
In concluzie, proteina RET mutanta nu mai necesita dimerizare pentru a deveni activa2.
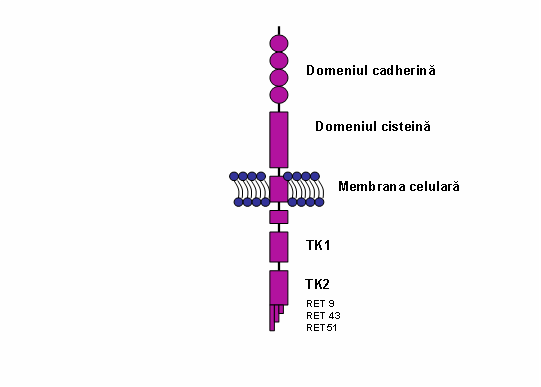
Fig. Reprezentare schematica a tirozin kinazei RET. Regiunea extracelulara este alcatuita din 4 domenii de cadherina si un domeniu bogat in cisteina; cele doua domenii tirozin kinazice sunt localizate in regiunea intracelulara (TK1 si TK2). Izoformele RET sunt indicate RET9, RET43 si RET51. (Adaptare dupa de Groot et al. 2006)
Recomandari pentru testarea genetica
-confirmarea diagnosticului la persoanele cu suspiciune clinica;
-testarea in scop predictiv a membrilor adulti asimptomatici din familiile cu risc crescut (imediat dupa nastere in cazul copiilor cu risc de MEN 2B si inainte de 5 ani pentru copii cu risc de MN 2A si CMTF); conform ASCO (American Society of Clinical Oncologists) MEN 2 este considerat un sindrom bine definit de cancer ereditar la care testarea genetica face parte din management-ul standard al membrilor cu risc crescut din familiile afectate;
-diagnosticul prenatal, in cazul sarcinilor cu risc crescut.
Pentru ultimele doua situatii este necesara identificarea in prealabil a mutatiei care a determinat boala in familie1;4.
Specimen recoltat – sange venos5.
Recipient de recoltare – vacutainer ce contine EDTA ca anticoagulant5.
Cantitate recoltata – 5 mL sange5.
Cauze de respingere a probei – folosirea heparinei ca anticoagulant; probe coagulate sau hemolizate4.
Stabilitate proba – 7 zile la 2-8ºC5.
Metoda – sunt disponibile doua modalitati de testare5:
- secventiere exoni 10, 11, 13, 14, 15 si 16;
- secventiere completa a genei RET.
Interpretarea rezultatelor
Spectrul mutatiilor genei RET in MEN2 este de 58 mutatii distincte, fiind afectati 19 codoni apartinand celor 7 exoni mentionati mai sus: 8, 10, 11, 13, 14, 15 si 16.
Sindromul MEN 2 prezinta o corelatie puternica genotip-fenotip:
● MEN 2A
Aproximativ 95% dintre familiile cu MEN 2A prezinta o mutatie RET la nivelul domeniului bogat in cisteina in exonii 10 si 11. Mutatia codonului 634, caracterizata prin substitutia cisteinei cu arginina in pozitia 634 (Cys634Arg), strict asociata cu aparitia feocromocitomului si/sau HPTP este intalnita in ~85% din cazuri6; mutatiile reziduurilor de cisteina de pe codonii 609, 611, 618 si 620 sunt responsabile de restul mutatiilor identificabile in exonii 10 si 11. Alte mutatii mai rare afecteaza codonul 804 de pe exonul 144.
● Cancerul medular tiroidian familial (CMTF)
Aproximativ 88% din familiile cu CMTF prezinta o mutatie RET. Aceste mutatii se produc la nivelul unuia din cele 6 reziduuri de cisteina (codonii 609, 611, 618, 620, 630 si 634), mutatiile codonilor 618, 620 si 634 fiind responsabile de 25-35% din cazuri. CMTF a fost de asemenea asociat cu mutatii ale domeniului intracelular al RET (la nivelul codonilor 768, 790 si 791 de pe exonul 13, codonii 804 si 844 de pe exonul 14 si codonul 891 de pe exonul 15)4;6.
● MEN 2B
Aproximativ 95% dintre persoanele cu fenotipul MEN 2B prezinta o mutatie punctiforma in domeniul tirozin-kinazic al genei RET la nivelul codonului 918 din exonul 16, care substituie metionina cu treonina (Met918Thr). La unii dintre pacientii care sunt negativi pentru mutatia Met918Thr a fost depistata a doua mutatie la nivelul codonului 883 din exonul 15 care inlocuieste alanina cu fenilalanina (Ala883Phe). Aceste doua mutatii sunt considerate a fi foarte agresive.
La pacienti MEN 2B au fost identificate si mutatii rare missens la nivelul exonilor 14, 15, 16 (codonii 804, 806, 904, 922)1;4.
In urma consensului de la cel de-al 7-lea Workshop MEN (1999) s-a efectuat o clasificare a mutatiilor RET in functie de riscul de CMT agresiv; aceasta clasificare este folosita in recomandarea varstei la care sa se efectueze tiroidectomia profilactica, pentru stabilirea necesitatii unui screening pentru feocromocitom si are valoare predictiva in ceea ce priveste fenotipul:
–mutatii de nivel 3 (asociate cu cel mai mare risc de CMT agresiv, fiind necesara o tiroidectomie profilactica in primele 6 luni de viata): mutatiile codonilor 883 si 918;
–mutatii de nivel 2 (este necesara o tiroidectomie profilactica inainte de varsta de 5 ani): mutatiile codonilor 609, 611, 618, 620, 630, 634;
–mutatii de nivel 1 (asociate cu cel mai mic risc de CMT agresiv, fiind necesara o tiroidectomie profilactica intre varsta de 5 si 10 ani): mutatiile codonilor 768, 790, 804, 891.
Toate subtipurile MEN 2 au o transmitere autozomal-dominanta:
-MEN 2A: aproximativ 95% dintre persoanele cu MEN2 identificate cu o mutatie RET au un parinte afectat; 5% prezinta fie o mutatie de novo a liniei germinale, fie o penetranta incompleta a alelei mutante;
-CMTF: prin definitie, pacientii cu CMTF prezinta numerosi membri ai familiei ce sunt afectati;
-MEN 2B: aproximativ 50% dintre persoanele afectate prezinta o mutatie de novo a liniei germinale si ~50% mostenesc mutatia de la un parinte; majoritatea mutatiilor de novo sunt de origine paterna.
Riscul fratilor de a mosteni mutatia depinde de statusul genetic al parintilor; daca un parinte prezinta o mutatie asociata cu boala riscul este de 50%; daca nu s-a putut detecta o mutatie la nici unul dintre parinti sunt posibile doua situatii: mutatie de novo la persoana afectata sau mozaicism al liniei germinale la un parinte. Fiecare copil al persoanei afectate are un risc de 50% de a mosteni mutatia.
Pacientii cu CMT sporadic (fara istoric familial) pot prezenta mutatii RET ale liniei germinale in 1-24% din cazuri si mutatii RET somatice ale codonului 918, care le confera o evolutie mai agresiva, in 30-67% din cazuri.
Mutatiile RET mai pot fi asociate si cu urmatoarele 2 afectiuni:
–boala Hirschprung: o afectiune a plexului enteric de la nivelul colonului asociata cu megacolon si constipatie cronica; 20-40% din cazuri sunt determinate de mutatii RET ale liniei germinale, insa in majoritatea cazurilor sunt implicati codoni diferiti de cei din MEN 2;
– carcinomul papilar tiroidian: in 40% din cazuri sunt prezente rearanjari somatice ale genei RET4.
Bibliografie
1. Abdelhakim A, Barlier A, Kebbou M, Benabdeljalil N, Timinouni M, Fechtali T, Roche C, El Antri S, RET genetic Screening in patients with medullary thyroid cancer: The Moroccan experience, In JCRT, 2009, 5 (3): 198-202.
2. De Groot JWB, Links TP, Plukker JTM, Lips CJM, Hofstra RMW, RET as a Diagnostic and Therapeutic Target in Sporadic and Hereditary Endocrine Tumors, In Endocrine Reviews, 2006, 27(5):535–560.
3. E. Tarcoveanu, Fl. Zugun. Neoplaziile endocrine multiple: de la diagnosticul genomic la chirurgia profilactica. In Jurnalul de Chirurgie, Iasi, 2007, Vol. 3, Nr. 1.
4. Georgia L Wiesner, Karen-Snow Bailey. Multiple Endocrine Neoplazia Type 2. Gene Reviews, 2010. www.ncbi.nlm.nih.gov. ReferenceType: Internet Communication.
5. Laborator Synevo. Referintele specifice tehnologiei de lucru utilizate 2010. Ref Type: Catalog.
6. Milos I, Frank-Raue K, Wohllk N, Maia AL, PusiolE, Patocs A, Robledo M, Biarnes J, Barontini M, Links TP, Willem de Groot J, Dvorakova S, Peczkowska M, Rybicki LA, Sullivan M, Raue F, Zosin I, Eng C, Neumann H, Age-related neoplastic risk profiles and penetrance estimations in multiple endocrine neoplasia type 2A caused by germ line RET Cys634Trp (TGC>TGG) mutation, In Endocrine-Related Cancer, 2008, 15 (4):1035 -1041.
Produsul a fost adăugat în coș
În plus, ai la dispoziție 30 de zile pentru a veni la recoltare.